Reaction optimization
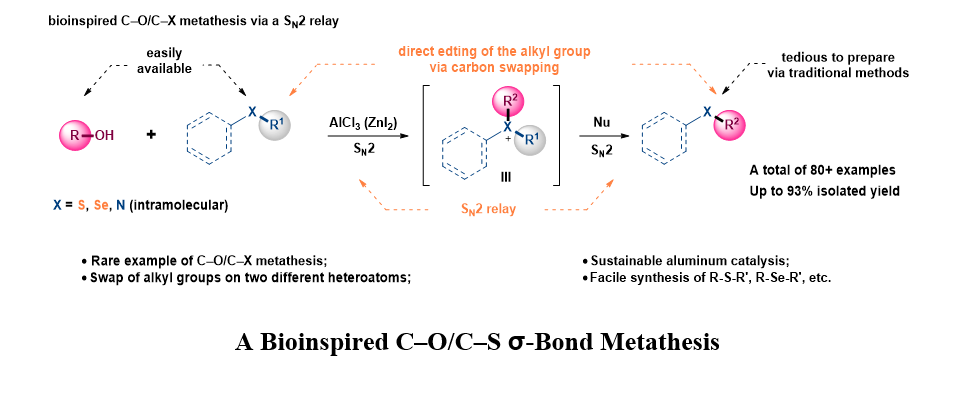
Although various sulfonium salt based alkylating reagents have been devised, their preparations and flowing reactions with nucleophiles are in separate steps that are not compatible with each other.30–31 To merge these two steps for a C–S metathesis is rather scarce with a highly reactive intermediate.32 We speculated that an SN2 relay process with high level of resemblance to that of SAM cycle is possible with a Lewis acid catalysis: activation of hydroxy group by a Lewis acid make it a good leaving group that can be displaced by a thioether, the Lewis-acid-bound hydroxy group could then act as nucleophile to react with the sulfonium intermediate to give a C–O/C–S metathesis product. To test this hypothesis, we started to evaluate the target C–O/C–S metathesis with 1-dodecanol (1a) and dimethyl sulfide (2a). As shown in Table 1, common Brønsted acids (entries 1–3) and Lewis acids (entries 4–5) failed to deliver the desired product in any noticeable amount. Encouraging results were obtained with FeCl3 (entry 6) and AlCl3 (entry 7), and a promising 41% yield of 3a could be obtained with an increased loading of AlCl3 (entry 9). Increasing the equivalency of 2a led to a slight decrease of the yields (entries 10–11). We then studied the possibility of adding a second Lewis acid (entries 12–17) as a cofactor and found that 50% ZnI2, although ineffective when acted alone (entry 4), can increase the yield to 60% when used in combination with AlCl3 (entry 17) and a good yield of 82% was obtained as one equivalent of ZnI2 was used (entry 18). When cyclohexane was used as the solvent, a slightly decreased yield of double C–O/C–S metathesis product 3a′ resulted in an improved selectivity for 3a (entry 19), while the virtual yield for the major is roughly the same. Other solvents (entry 20, and Table S2) and iodides were proved to be less effective as a cofactor (entries 21–22, and Table S2).
Synthetic scope
With the optimized reaction condition for the C–O/C–S σ-bond metathesis in hand, we next explored the substrate scope of this transformation (Fig. 2). Generally, primary aliphatic alcohols with variable length and aliphatic substituents all reacted with Me2S (2a) to give the corresponding alkyl methyl sulfides in moderate to excellent yields (3a–3e), a phthalimide moiety (3f–3g) and a second sulfide moiety (3h) were also well tolerated albeit with slightly decreased yield. Cyclic secondary alcohol was also a viable substrate, and the σ-bond metathesis product 3i could be obtained in an isolated yield of 70%. Other than Me2S, Et2S and Pr2S both reacted under the standard condition to give 3j and 3k in good isolated yields. When unsymmetrical thioethers 2d and 2e were used, the second SN2 reaction of the C–O/C–S metathesis preferred to occur at the less hindered methyl group to give 3b′ and 3b′′ as the major products (Fig. 2, inset). We next evaluate the metathesis reaction of Me2S with more challenging aromatic alcohols. The oligomerization of benzyl alcohol would be depressed, and the corresponding C–O/C–S σ-bond metathesis product 3l was obtained in 52% isolated yield. Variable substituents on benzyl alcohols were all well tolerated (3m–3p). 2-phenylethanols with various substituents (3q–3zz) and 2-naphthylethanols (3bb–3cc) also reacted under the standard condition to give the corresponding C–O/C–S σ-bond metathesis products. 3-phenylpropanols and 4-phenylbutanols have potential competitive intramolecular Friedel-Crafts reaction. Luckily, the products 3dd–3ee and 3ff–3gg could still be obtained in moderate to good yields. Finally, allylic alcohols such as cinnamon alcohols usually give complicated mixture due to dehydration and Friedel-Crafts reaction. However, when substituted with an electron-withdrawing group to depress the Friedel-Crafts reaction, C–O/C–S metathesis product 3hh could be obtained in moderate yield.
Encouraged by this initial success, we next turned our attention to the more challenging aryl methyl thioether (Fig. 3, upper half). The metathesis of the C–S bond for the synthesis of more complicated homologs was only possible with transition metal catalysis that relies on ligand exchange with a second thiolate15–16 or with the formation of a highly reactive aryne intermediate32 or over an inoinc liquid.33 To our delight, under the optimized reaction condition, simple aliphatic alcohols (4a–4d), alcohol bearing a phthalimide moiety (4e), and aromatic alcohols (4f–4h) can react with commercially available methyl phenyl thioether to give the alkyl transposition product, which is usually cumbersome to obtain with previous methods. A substrate scope study with 2-phenyl alcohol showed that various substituents, including alkyl (4i), electron-withdrawing (4j), electron-donating (4k), as well as halogens (4l–4p) on the aromatic thioethers are all well tolerated. methyl(naphthalen-2-yl)sulfane is also a viable substrate to deliver the product 4q in 63% isolated yield. Ethylene glycol and 1,3-propanediol underwent a double C–O/C–S metathesis to give 4r and 4s in 46% and 35% isolated yield, respectively. Due to the similarity of the same group element, aromatic selenoethers can also react with various alcohols under the standard condition to give the unusual C–O/C–Se metathesis products 4t–4w in good isolated yields. To further justify the synthetic potential of the target C–O/C–X σ-bond metathesis reaction, we evaluated the intramolecular variants for the syntheses of saturated heterocycles, which could present an important complement to the renowned ring-closing olefin metathesis (Fig. 3, lower half). Fortunately, ring-closing C–O/C–S metathesis gave access to cyclic thioethers of diversified electronic properties (6a–6i). The reduced yield of 6f might be due to the basicity of the cyano group that influences the combination of ACl3 and ZnI2, and a double C–O/C–S σ-bond metathesis gives 6j in synthetically useful 38% yield. In analogy, ring-closing C–O/C–O σ-bond metathesis24–27 could also be achieved for substrates with ethers instead of thioethers (6k–6o). Surprisingly, an unprecedented ring-closing C–O/C–N σ-bond metathesis could be implemented under the standard condition to give highly valuable cyclic amines (6p–6t), although an intermolecular variant was still elusive with the current protocol. A brief substrate scope study showed that substituents of various electronic properties were well tolerated.
Synthetic applications
Two gram-scale reactions for the synthesis of 3a and 4h were carried out, respectively, and yields comparable to those of the small-scale reactions were obtained. In order to further test the synthetic potential application of this methodology, we successfully realized the upgrading of Me2S via two successive C–O/C–S σ-bond metathesis reactions that convert easily available Me2S to more advanced symmetrical (3bb) and unsymmetrical (3bc) thioethers in synthetically meaningful isolated yields that were otherwise much more tedious to obtain (Fig. 4a). (3-bromophenyl)(methyl)sulfane (7) is a key synthetic intermediate to the dopamine D2 antagonist Priodopidine (8), which has neuroprotective effects on mitochondrial function, ion channel function, as well as ER stress in various experimental systems of amyotrophic lateral sclerosis (ALS), Parkinson’s disease and Huntington’s disease.34 By applying the title C–O/C–S σ-bond metathesis reaction, we can convert 7 to 7′ or 7′′ in a single operation that could be further elaborated to analogs with an apolar (8′) or polar (8′′) side-chain respectively (Fig. 4b).
Mechanistic investigations
We then carried out a series of control experiments to possibly reveal the mechanism underlying this C–O/C–S σ-bond metathesis reaction. (Fig. 5) Firstly, we analyzed the reaction mixtures performed at various temperatures and stopped at various stages by gas chromatography (GC). In all of these cases, other than σ-bond metathesis product 3a (if the reaction temperature is appropriate), only dehydrated product 1a-1 was detected as a side product while halogenated byproduct 1a-2 or 1a-3 that might be formed in these Lewis acids were not detected (Fig. 5a). The same analysis was also done without the presence of Me2S. However, 1a-2 or 1a-3 was still not detected, although a new ether 1a-4 was detected, excluding the possibility of forming 1a-2 or 1a-3 as highly reactive intermediates that were quickly consumed in the following step. When alkene 1a-1 was subjected to the standard reaction condition, no 3a was formed, ruling out the possibility of 1a-1 as an intermediate in the catalytic cycle (Fig. 5b, Eq. 1). On the other hand, when ether 1a-4 was submitted to the reaction condition, 3a could be obtained in significantly reduced yield with significant 1a-4 remaining unreacted. This information, together with the fact that no 1a-4 was detected in a standard reaction, suggests that 1a-4 is less likely to be in the catalytic cycle (Eq. 2).33 Although 1a-2 and 1a-3 were not detected in the reaction mixture when 1a-2 was submitted to the standard reaction condition, a reduced yield of 3a was obtained and no 3a in the absence of ZnI2 (Eq. 3), in contrast, 1a-3 gave higher a yield of 3a in the absence of ZnI2 while its presence depressed the reaction (Eq. 4). These results pointed to a complicated role of ZnI2 and multiple possible reaction pathways for the first SN2 of this relay. A kinetic isotope effect experiment with 2g and 2g-d3 showed an obvious normal secondary isotope effect for both parallel experiments (kH/kD = 1.33, Fig. 5c) and competition experiment (kH/kD = 1.27, Figure S11). This suggested that the nucleophilic attack of the sulfonium intermediate at the methyl group (Second SN2, sp3 to sp2) is likely to be the rate-determining step. Finally, experiments with various radical trapping agents successfully delivered the product in good yields (Fig. 5d), except for TEMPO, the basicity of which might be incompatible with the mixture of Lewis acids. No trapped radicals could be detected for all trapping agents tested, ruling out the possibility of a radical pathway, a mechanism that is common in SAM involved biological process.35
DFT Calculations
Parallel to experimental studies, density functional theory (DFT) calculations were conducted to provide further mechanistic insights into the title reaction. Four different types of substrates, forming products 2d, 3l, and 4h, 6a, were considered using AlCl3 as the catalyst. The Gibbs energy diagram for the intermolecular C-O/C-S σ-bond metathesis production of 2d (left) and ring-closing C-O/C-S σ-bond metathesis production of 6a (right) are shown in Fig. 6, while others are displayed in the Supporting Information. The calculations suggested a stepwise SN2 relay mechanism for both reactions. In the first step, the sulfur performed the nucleophilic attack on the carbon center, coupled with the departure of the hydroxide, which is stabilized by the Lewis acid AlCl3. This leads to the generation of an ion-pair intermediate (Int1A or Int1B). Subsequently, the hydroxide attacks the methyl group of Int1A or Int1B, concomitant with the S-C bond cleavage. The barriers for the first SN2 were computed to be 24.3 kcal/mol and 22.8 kcal/mol, respectively, while the barriers for the second SN2 were 26.3 kcal/mol and 24.8 kcal/mol, respectively, suggesting that the second SN2 step should be rate-limiting for both reactions, agreeing with the kinetic isotope effect experiments shown in Fig. 5c. Overall, the ring-closing C-O/C-S σ-bond metathesis has a more favorable energetic profile compared to the intermolecular variant. Similar results could be found for the other three substrates, with the second step being rate-limiting, and the barriers are around 25 kcal/mol (see the supporting information).
We have reported in this article a biomimetic C–O/C–S σ-bond metathesis reaction between alcohols and thioethers. This method allows facile access to cumbersome thioethers via direct editing of easily available ones by swapping one alkyl group with that of an alcohol. This protocol could be extended to C–O/C–Se σ-bond metathesis for the synthesis of selenoethers. When rendered intramolecular, cyclic ethers, thioethers, or amines could be synthesized by direct C–O/C–X σ-bond metathesis. Mechanistic studies, as well as DFT calculations, agreed with an “SN2 relay” mechanism that showed a high level of resemblance to that of the natural SAM cycle. We anticipate that this methodology will enrich the toolbox of synthetic chemists for the construction of chemically and biologically important thioethers, cyclic ethers (thioethers), and cyclic amines. From a broader perspective, we anticipate this bioinspired design of C–O/C–X metathesis will provide insight for developing novel cross-metathesis reactions involving inert C(sp3) –X as well as C(sp2) –X bonds.
{kind=link}