Quantitative structure-activity relationships (QSAR) modeling is an in silico methodology aimed at predicting the physical or biological properties of small molecules. A QSAR analysis generally attempts to find correlations between an experimental biological activity and molecular descriptors, either calculated from the chemical structure of the compound or experimentally obtained [1, 2]. QSAR modeling has diversified and evolved from its application to small series of similar compounds, using relatively simple regression methods, to the analysis of much larger datasets spanning thousands of molecules, using various statistical techniques [3]. Continuous improvements have allowed QSAR modeling to be employed in the chemical, medical, and pharmaceutical industries and by government institutions [4].
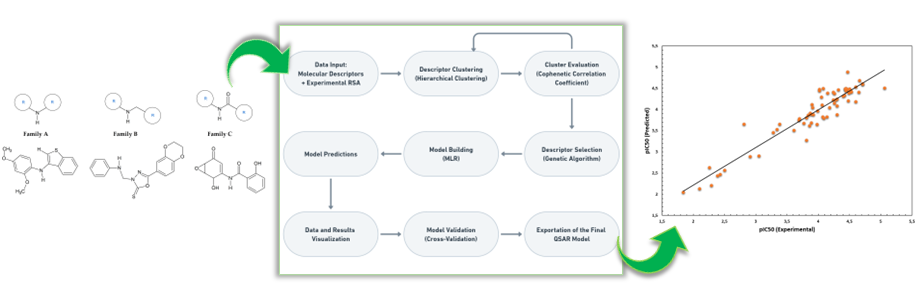
A series of steps are typically needed to perform a QSAR analysis: chemical data preparation (literature search for compounds with an IC50 value defined experimentally by comparable biological assays), dataset splitting, molecular descriptor calculation, descriptor selection, mathematical model building and model validation (Fig. 1).
Currently, many software tools are available, and typically, each software performs one or a small number of steps needed to build a QSAR model [3, 5] or is a commercial software that is not readily available for many researchers. PyQSAR is a free, open-source software that performs several steps of QSAR model development [6], while OCHEM is another free-for-academic use web-based platform that calculates a large number of molecular descriptors [7]. The calculated descriptors range from 1D descriptors, based on the physicochemical properties or molecular formula of the compounds, to 2D descriptors, calculated from a 2D representation, and 3D descriptors obtained from a 3D representation of the compounds [8]. Recently, a workflow was presented by Mansouri et al. (2024) based on KNIME software that automated the initial steps of QSAR modeling, specifically chemical library structure preparation [9]. Additionally, a framework for QSAR model building was published by Kausar et al. (2018); however, the number of available molecular descriptors is limited, and extensive knowledge of the KNIME platform is needed [10].
Reactive oxygen species (ROS), which are commonly generated as byproducts of chemical reactions within the human body, inflict harmful effects on living cells. Oxidative stress occurs when the body's natural antioxidant defense system cannot effectively neutralize the free radicals produced within the body [11–13]. The antioxidant activity is a biological property of small molecules that have been widely studied and is related to the treatment and prevention of several diseases, including inflammatory diseases (e.g., rheumatoid arthritis), diabetes and neurodegenerative diseases (e.g., Alzheimer's disease) [14, 15]. Several antioxidants have been developed and used in the cosmetic, pharmaceutical, and food industries [16]. Due to its importance, several antioxidant QSAR studies of different compound classes have been reported, consubstantiating the applicability of this type of study [17–20].
A study by Goya Jorge et al. (2016) built an antioxidant activity QSAR model using a library of 1373 compounds. As the experimental variable, the radical scavenging activity (RSA) was obtained using the DDPH (2,2-diphenyl-l-picrylhydrazyl) method. To get the QSAR model, Goya Jorge et al. (2016) used a neural network method called multilayer perceptron (MLP) while using DRAGON® software to calculate the molecular descriptors [17]. In a previous study, a library of 26 di(hetero)arylamine and amine derivatives was also used to prepare an antioxidant QSAR model. To build the QSAR model, Abreu et al. (2009) used the partial least squares projection of latent structures (PLS) method and DRAGON® software to calculate the molecular descriptors [18]. The antioxidant activity of each compound was experimentally determined using the DPPH method, which assesses each compound's free RSA, and the results are presented as IC50 values. The statistical performance was outstanding, with an R2 value of 0.881 and a Qext2 value of 0.843. The di(heteroaryl) amines and amides are thus promising scaffolds for developing new compounds with potent antioxidant activities [18]. Another study by Zhan et al. (2017) prepared a 3D-QSAR model to analyze the relationship between the antioxidant activity of 15 amine derivative compounds as additives of trimethylolpropane trioleate lubricating oil [20].
We present a complete methodology for preparing QSAR models using free and open-source software tools, from chemical library preparation to calculating and selecting molecular descriptors to QSAR model building and validation. As an example of an application of the methodology, an antioxidant QSAR model was prepared using a library of 70 di(hetero)aryl amines or amides. The main tools used were the OCHEM platform to calculate the molecular descriptors while PyQSAR was used to build the QSAR model. The developed antioxidant QSAR model presents excellent standard and cross-validated statistical parameters and can be used to predict and guide the synthesis of new di(hetero)aryl derivatives with improved antioxidant activities. We describe in detail the methodology used and present an easy-to-follow step-by-step protocol for developing similar QSAR models for different biological activities that may be of interest to other researchers (Additional file 1).
{kind=link}