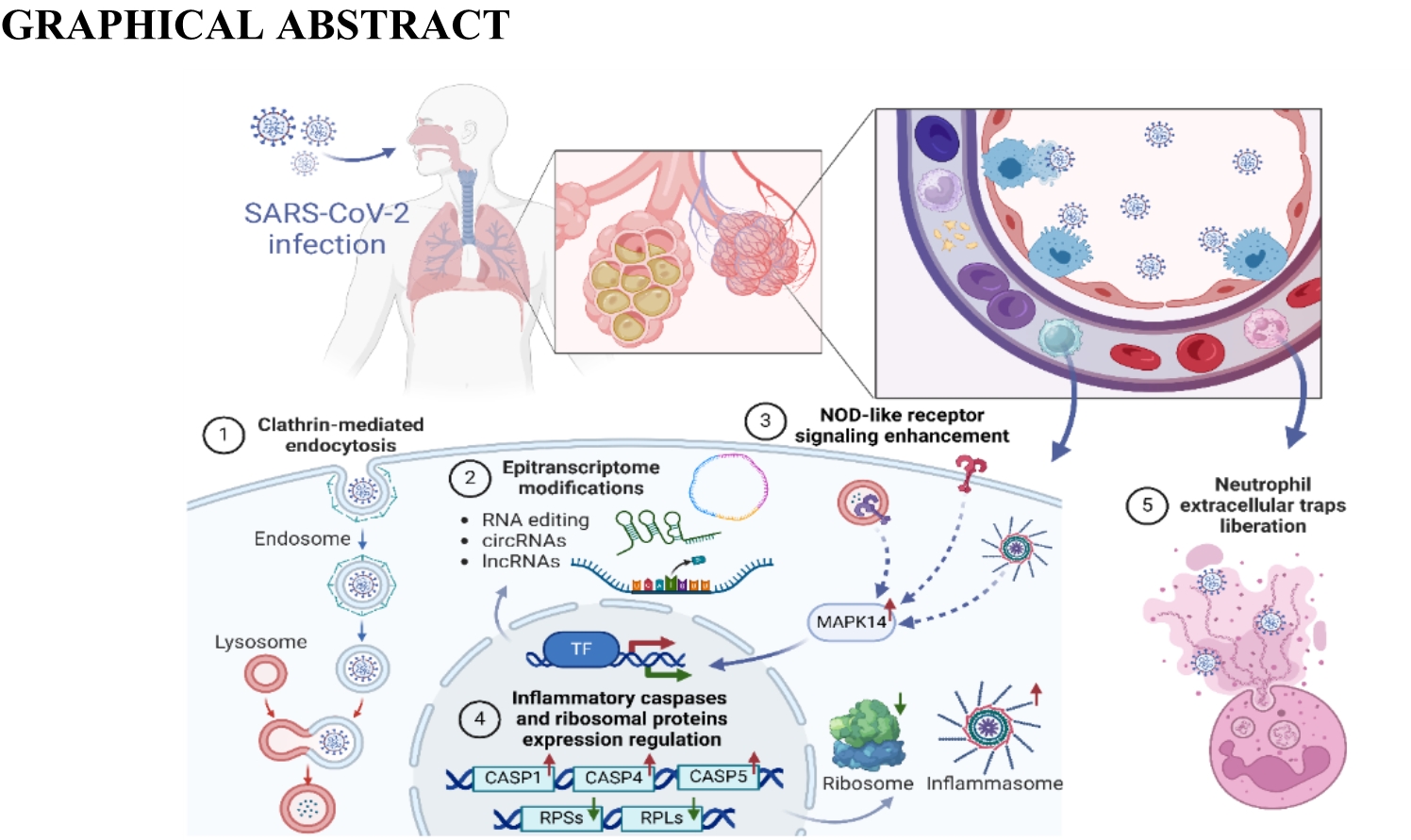
Since the beginning of the pandemic, some attempts have been made to investigate the transcriptomic profile of peripheral blood samples of the SARS-CoV-2-infected patients. A whole-blood RNA expression signature for COVID-19 patients of Asian origin performed by Kwan et al. [18] revealed 135 protein-coding DEGs. Some of these genes were also present in datasets from Respiratory Syncytial Virus (RSV) and Influenza, but predominantly they were unique to COVID-19. The revealed protein pathways belonged mostly to generic responses to viral infections, such as type-1 interferon and inflammatory responses, as well as apoptosis by the p53-associated pathway, but also included some unique pathways such as viral carcinogenesis. Another study investigating the COVID-19 blood transcriptome revealed a lot of significantly altered genes associated with antiviral defense, mitotic cell cycle, type-1 interferon signaling, and severe viral infections [23]. Wargodsky et al. [59]using RNA-seq of the blood samples of COVID-19 patients revealed, that SARS-CoV-2 infection and the severity of COVID-19 were associated with up to 25-fold increased expression of neutrophil-related transcripts, such as neutrophil defensin 1 (DEFA1), and up to 5-fold reductions in T cell-related transcripts such as the T cell receptor (TCR) [59]. Moreover, it has been also found that SARS-CoV-2 proteins directly interact with the host RNAs to suppress global mRNA splicing and translation, affect signal recognition and interfere with protein trafficking to the cell membrane. Dysregulation of these crucial functions is supposed to suppress the interferon response to viral infection [27]. In the current research, we analyzed the whole blood transcriptome of COVID-19 patients using RNA-seq and Nanopore validation. The GO, REAC and KEGG annotations indicated that SARS-CoV-2 infection resulted in profound changes in the expression of several genes previously assigned to: Coronavirus disease—COVID-19, SARS-CoV-2-host interactions, or response to the virus. Additionally, several biased genes revealed in the infected patients belonged to the NOD-like receptor signaling pathway (KEGG).
SARS-CoV-2 gains entry into the host cells through the ACE2 receptor, expressed in various organs like the kidney, lung, and heart [60]. After a virus enters the host body, many biomolecules are engaged in its internalization into host cells. The PICALM is involved in clathrin-mediated endocytosis, which plays a crucial role in the internalization of SARS-CoV-2 into lung epithelial host cells [61, 62]. Interestingly, our results revealed the multiple intronic ASEs that co-occurred with exonic circular self-splicing within this gene in SARS-CoV-2 infected group suggesting that these molecular events play a crucial role in PICALM mRNA maturation and transcription during COVID-19 disease. Thus, our results support pointing PICALM as one of the targets for new therapies against virus-induced suppression of interferon-induced innate immunity, promotion of viral replication and subversion and/or evasion of antiviral immune surveillance [63]. Fernbach et al. [64] discovered that overexpression of RABGAP1L can cooperate with interferons against a large spectrum of viruses, including Influenza [64]. Depending on the infection stage, RABGAP1L high expression level disrupts the virus-host membrane fusion, the main path of pathogen entry. Otherwise, SARS-CoV-2 virus seems to be resistant to the inhibitory character of RABGAP1L. Our results supported this thesis, indicated by the underexpression of RABGAP1L and the silencing nature of the intron retention event (IncLevDiff = 0.16) observed in all COVID-19 patients.
In the blood of COVID-19 patients, the gene's expression revealed significant changes, with implications for antiviral defense, the mitotic cell cycle and type I interferon signaling. Some of the biased genes found in the experiment have been previously linked with COVID-19. Among interesting gene signatures identified in the current study, the upregulated immune biomarker Interferon Alpha Inducible Protein 27 (IFI27) seems to be an important player in the immune response to SARS-Cov-2 infection. It has been found that the upregulated IFI27 is associated with high numbers of the SARS-Cov-2 virus particles and may be considered an early predictor for COVID-19 outcomes [65]. Its importance in the COVID-19 progression is associated with its primary function as an IFN-stimulated gene and modulatory potential towards immune response [66]. The immune response triggers the activation of genes related to neutrophil mechanisms, secretory granules, and neutrophil extracellular traps (NET) [23]. NETs, however, are involved in the induction of myocardial infarction and stroke. Patients with severe COVID-19 suffer from complications such as myocardial infarction and stroke with pathological signs of NETs [67]. NETosis is accompanied by lamin B1 degradation and nuclear membrane rupture [68]. The laminin B1 breakdown was most probably the cause of the observed upregulation of the Lamin B1 (LMNB1) gene in SARS-CoV-2 infected group in the current data. The current data revealed that Alpha Kinase 1 (ALPK1) was not only upregulated but also alternatively spliced in the COVID-19 group. The overexpression of this gene has been previously found in the peripheral blood mononuclear cells of rhesus monkeys infected with SARS-CoV‐2 [69]. Most possibly the upregulation of ALPK1 increases the production of inflammatory cytokines and chemokines during the immune response [70]. Additionally, the present findings revealed that SARS-CoV-2 infection is followed by the upregulation of Mitogen-Activated Protein Kinase 14 (MAPK14), which encodes a member of the p38 subfamily, known to regulate inflammatory cytokines and exerts an essential role in inflammatory acute lung injury [71]. Moreover, MAPK14 is one of the core targets of baicalin action to treat cytokine storm [72]. MAPK14 was identified as a high-ranking gene that may be a potential pharmacological target, fulfilling the requirements for a fast and safe drug for COVID-19 [73, 74]. Further, the KEGG analysis performed in the current research revealed that several biased genes, including MAPK14 were engaged in the NOD-like receptor signaling pathway. Interestingly, Qiu et al. by using the bioinformatics approach uncovered the critical role of the NOD-like receptor signaling pathway in COVID-19-associated multiple sclerosis syndrome [75]. The intracellular NOD-like receptors are crucial components of innate immune response and inflammation [76], and their activation among others triggers the formation of caspase-1 (CASP1) mediated canonical inflammasomes and noncanonical inflammasomes involving CASP4 and 5 [77]. In the blood of COVID-19 patients, the current research revealed the upregulation of CASP1, CASP4 and CASP5, suggesting activation of both these pathways. Indeed CASP1 has been previously considered an important mediator of exaggerated inflammatory response in COVID-19, and it has been proposed that inhibition of the CASP1 expression, for instance by minocycline, may prevent the cytokine storm inflammation [78]. Indeed it has been revealed that the upregulation of caspases, by NOD-like receptors, is responsible for the exacerbation of COVID-19. Upon infection, SARS-CoV-2 induces endotheliitis, leading to vascular dysfunction and subsequent tissue damage [79]. This process involves the inflammation and activation of endothelial cells, resulting in the disruption of the endothelial barrier function. The lung, in particular, is highly impacted due to its significant proportion of endothelial cells [79, 80], thus, respiratory and cardiovascular disorders have emerged as prominent threats to the progression of this disease [81]. In COVID-19 patients the present study revealed the upregulation of Adrenomedullin (ADM), a gene that has been shown to play a key role in regulating vascular permeability and endothelial integrity associated with severe infection [82] and COVID-19 induced endotheliitis [83]. The above changes, marked by increased ADM plasma levels, were triggered by proinflammatory cytokines and the degradation of the basement membrane by matrix metalloproteinases that are released by activated endothelial cells [84]. It has also been proved that ADM expression in whole blood was higher in patients with COVID-19 than in patients suffering from other respiratory infections and its elevation was associated with the severity of COVID-19 [85]. Data showed that high plasma bioactive-ADM (bio-ADM) concentration was elevated in non-survivors compared to survivors [84]. Thus, findings highlight the potential of bio-ADM as a promising biomarker for the early risk assessment of critically ill patients suffering from COVID-19 [84].
Current throughput transcriptomic analysis revealed that SARS-CoV-2 has a significant impact on the expression of multiple genes encoding the ribosomal proteins (RPs), including ribosomal protein L (RPL) and ribosomal protein S (RPS). The RPs' canonical ribosomal functions are involved in extra-ribosomal pathways including activation of p53-dependent or p53-independent response to stress, resulting in cell cycle arrest and apoptosis [86, 87]. Some of the RPs were previously linked with an immune response to a viral infection, and exemplary Ribosomal Protein S23 (RPS23) was previously reported among down-regulated genes whose expression was associated with SARS-CoV-2 viral load [88]. The under-expression of RPS23 has been also detected in blood samples taken from COVID-19 patients in the current research. It has been found that in blood B lymphocytes, RPS23 plays a role in ribosome assembly and protein translation, which may be related to antibody production by B cells [89]. Moreover, RPS23 has also been reported to be a new peptide that can recognize and kill potential pathogens [90] and plays an important role in immune signaling [91]. RPS23 was also identified as a gene that differentiates in immune responses to COVID-19 vaccination strategies [92]. Moreover, another downregulated gene Ribosomal Protein S8 (RPS8) revealed in the present study in COVID-19 patients, is listed among key genes for the advancement of SARS-CoV-2 infection and is suggested as a potential novel biomarker that may serve as a diagnostics or therapeutic target for this infection [93]. The RPSs and RPLs genes, such as RPL3, RPL7, RPL23A, RPL34, RPS3A, RPS8 and RPS23 were listed among the top-ranked genes connected with postoperative systemic inflammatory dysregulation [86], and they were also detected in multiple analyses performed during current research in SARS-CoV-2 infected patients. Obtained results indicate that multiple RPSs and RPLs genes differentiate not only in terms of expression but also manifest biases in alternative splicing (RPLS15 and RPS24) and ASE frequency (RPL37 and RPS6KA1). Additionally, it is important to note that most of the RPSs and RPLs displayed differences in datasets obtained using RNA-seq as well as Nanopore sequencing. The joint functional analysis points that RPs mostly affected pathways such as coronavirus disease—COVID-19, SARS-CoV-2-host interactions, response to the virus and NOD-like receptor signaling pathway. Therefore, considering intra- and extra-ribosomal functions it could be hypothesized that biased expression of the RPs in the context of the COVID-19 severity may be considered as a factor contributing to the altered immune competence. Intriguingly Alfi et al. revealed that the significant impact of SARS-CoV-2 on pathways engaged in translation regulation involves not only the repression of ribosomal genes, such as RPL5, RPL7, EEF2, RPS6, RPS4X and EEF1A1 [94]. Our results also strongly depict the characteristic manner of SARS-CoV-2 infection strategy, reflected not only by the downregulation of EEF1A1 but also by the repression of genes responsible for the translation of host proteins such as RPL5, RPL7, EEF2, RPS6, RPS20, RPL31, RPL19). This finding could be related to the multifaceted strategies employed by SARS-CoV-2 to suppress host protein synthesis [94]. In blood T cells, Eukaryotic Translation Elongation Factor 1 Alpha 1 (EEF1A1) is responsible for the GTP-dependent binding of aminoacyl-tRNA to the ribosome [92, 95]. The N protein of the SARS-CoV-2 binds to EEF1A to induce its aggregation, thereby inhibiting the synthesis of the host protein [96]. The knock-down of EEF1A1 has been shown to significantly impair the virus infection cycle, therefore, the downregulation of EEF1A1, observed also in the investigated COVID-19 patients, could be the result of an innate cell strategy to deprive SARS-CoV-2 of key support for RNA replication [97]. EEF1A1 as a translation initiation factor essential in viral infections can be used as drug targets for the treatment of COVID-19 and has a therapeutic potential [98]. This assumption can be supported by the fact that plitidepsin and ternatin4 (which are inhibitors of EEF1A) have a potential preclinical effect on SARS-CoV-2 [96]. Furthermore, the present results revealed not only the upregulation but also allelic-specific expression in the Makorin Ring Finger Protein 1 (MKRN1) gene in the COVID-19 group. The protein encoded by MKRN1 belongs to the CCCH-type zinc finger proteins which exhibit the capacity to interact with virus RNA and control its replication. The changes in MKRN1 expression during the SARS-CoV-2 infection most likely arise since viruses intervene with RNA metabolism [99]. MKRN1 is currently considered an anti-viral protein as it has been found to inhibit virus replication, by inducing ubiquitination and proteasomal degradation of its capsid protein [100, 101]. A specific intronic variant of this gene may be a novel marker in the molecular response to the COVID-19 infection. The MKRN1 downregulation observed in the present study was unusual, which suggests that the antiviral response in the SARS-CoV-2 patients was disturbed.
Recent research revealed that the host may repress virus infection by epigenetic biomolecules [102], such as ATRX Chromatin Remodeler (ATRX) gene, which regulates chromosomal instability and prevents DNA damage [103]. Our research revealed that ATRX expression can be modulated by circular RNA (exonic) and splicing events (RI and A3SS) differentiations within their transcripts. In human cells, two enzymes ADAR and APOBEC are involved in the editing of RNA [104]. Recent research revealed that the protein encoded by another upregulated gene revealed in COVID-19 patients, namely Apolipoprotein B MRNA Editing Enzyme Catalytic Subunit 3A (APOBEC3A) plays a crucial role in restricting viruses belonging to the Coronaviridae family [105]. The APOBEC3 proteins with inhibitory capabilities link the N protein, essential for the formation of the viral ribonucleocapsid, crucial in virus replication [105]. Viruses have developed intricate strategies to facilitate their replication, relying entirely on the host's translational apparatus for synthesizing proteins. The SARS-CoV-2 infection triggers host kinases which induce phosphorylations used by viruses for their better survival and further mutations. Thus, host cells respond to viral infection by inhibiting protein synthesis and disrupting mRNA transport [74]. Therefore, many proteins in SARS-CoV-2-infected individuals decrease in abundance relative to uninfected controls, in particular, this refers to the host phosphoproteome [106]. The present data revealed several genes with ASE sites, including Adenosine Deaminase RNA Specific (ADAR). The allele specification within the 3'UTR region suggests that ADAR expression may be regulated by different mechanisms in both research groups. This makes a big impact on the basic regulation of the RNA editing process dependent on ADAR functionality. In human cells, ADAR proteins are involved in RNA editing by introducing signature point substitutions cytosine to uracil (C-to-U) and adenosine to inosine (A-to-I) into the viral genome [104]. RNA of bronchoalveolar fluids from patients infected with SARS-CoV-2 was enriched in nucleotide changes (C-to-U and A-to-I) that might be indicative of the host deaminases involvement in the RNA editing, suggesting ADAR-mediated editing activity in the SARS-CoV-2 infection [107]. Extensive C-to-U and A-to-I nucleotide substitution patterns have been observed upon SARS-CoV-2 genome analysis. A recent study showed that ADAR1 was among the most upregulated genes in pulmonary alveolar type II cells of patients infected with SARS-CoV-2 [108]. Similarly, ADAR1 expression was increased in the peripheral blood cells of patients with severe COVID-19 compared to those with mild disease [109]. Upregulation of ADAR1-induced RNA editing in patients with COVID-19 could further propagate the inflammatory response by increasing the stability of proinflammatory transcripts [110, 111]. Perturbations of the epitranscriptome of COVID-19 patients indicate that mechanisms including those derived from RNA modifications and non-coding RNAs may play a contributing role in the pathogenesis of COVID-19 [112]. This observation holds significant implications for advancing novel therapeutic approaches in the treatment of COVID-19. Among the different strategies, the one that suggests molecular mechanisms introducing genomic alterations into the SARS-CoV-2 genome, like an increase in the involvement of deaminases belonging to ADAR in the editing of the SARS-CoV-2 RNA, is worth pointing out. This approach may additionally diminish the already low cytosine presence within the viral genome, resulting in compromised integrity and ultimately neutralizing the virus [113].
Alternative splicing of many gene targets can bring an explanation of some protein degradation as a consequence of open reading frame shortening. The current data unveiled the dynamic changes in the number of alternatively spliced genes during COVID-19 disease progression. The Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2 (EIF2AK2) is a well-known gene encoding an antiviral protein that mediates translation inhibition of host and viral mRNAs through phosphorylation. SARS-CoV-2 infection causes phosphorylation at the important S33 residue of antiviral kinase EIF2AK2 [74]. Our study revealed that EIF2AK2 has been alternatively spliced (A3SS type) in COVID-19 patients, which most probably deteriorates the antiviral function of the protein coded by this gene, and makes it easier for SARS-CoV-2 to replicate. Partial inclusion of intron sequence in EIF2AK2 transcripts may cause unexpected translation termination and in consequence function deactivation. A similar effect may result from the alternative splicing in 2'-5'-Oligoadenylate Synthetase (OAS1) observed in our study in the infected patients. The OAS1 was previously described as a SARS-CoV-2 strong restriction factor capable of inhibiting viral infection [114]. OAS1 isoforms have been shown to differ in their antiviral activities [115]. The canonical isoform of OAS1 exhibited restriction of infection. The inactivation of OAS1 catalytic activity completely ablated its antiviral effects, suggesting that the mechanism of OAS1-mediated SARS-CoV-2 restriction acts through downstream RNase L activation which degrades cellular and viral RNA [114]. OAS1 genetic variants were linked to infection and excessive morbidity in the SARS-CoV outbreak [116, 117]. More recently, a genome-wide association studies (GWAS) associated single nucleotide polymorphism (SNPs) within OAS1 loci with COVID-19 mortality [118]. Our results revealed that OAS1 may be regulated by alternative splicing events, especially intron retention splicing that cause shorter OAS1 variants, dominant in the COVID group. Interesting changes were detected within the BAZ1A gene. In this case, the three expression levels indicated significant modulations: alternative splicing events, circular RNA inverse ligation and variant-specific expression. BAZ1A controls DNA accessibility and chromatin remodeling [119] and may have a role in transcription regulation during SARS-CoV-2 infection. The observed co-occurrence of alternative splicing with circular self-splicing events supports the hypothesis that these molecular processes may be crucial to the regulation of BAZ1A mRNA during COVID-19 infection. Moreover, in SARS-CoV-2 infected host cells, our study identified the alternative splicing and differentially expressed profiles in one of the top expressed markers and drug targets for COVID-19 disease, namely ADP ribosylation factors like GTPase 4A (ARL4A) gene [120].
{kind=link}