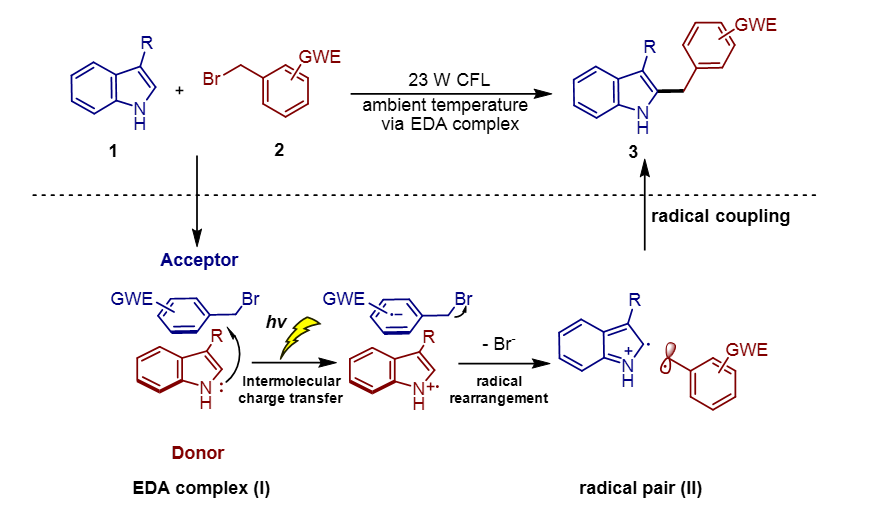
EDA complexes formed by 3-methyl-1H-indole derivatives and substituted benzyl bromide.
To determine the effect of different positions and types of substituents on the benzyl bromide derivatives and indole derivatives comprising the EDA complexes on the magnitude of the binding energies of their constituent EDA complexes. We considered and calculated the EDA complexes of benzyl bromide derivatives substituted at different positions of the electron-withdrawing group (e.g., nitro, nitroso, cyano, or aldehyde) with 3-methyl-1H-indole derivatives; EDA complexes of benzyl bromide derivatives substituted at different positions of the electron-donor group (e.g., methyl or methoxy) with 3-methyl-1H-indole derivatives; Finally, the EDA complexes of nitro-substituted 3-methyl-1H-indole derivatives at different positions and in different amounts with methoxy-substituted, unsubstituted and methoxy-substituted benzyl bromide. In the process of obtaining the bonding energies of the complexes mentioned above, we chose the zero-point energy and compared the bonding energies of the mentioned EDA complexes (for details, see ESI, † Fig. S1-S5).
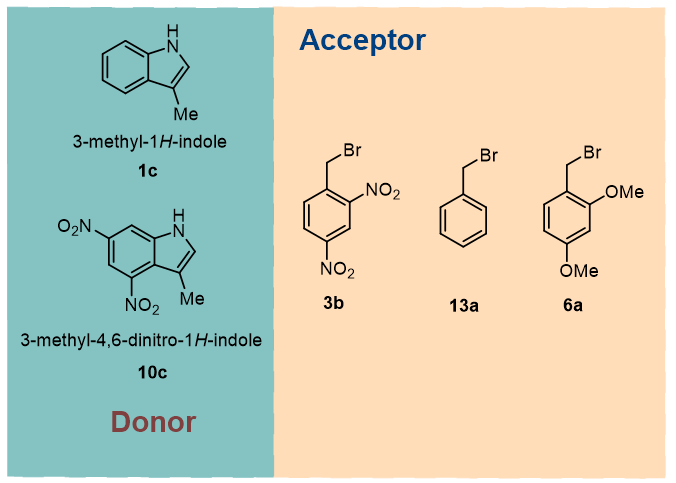
All electron-withdrawing group substituted benzyl bromide derivatives have lower binding energies than unsubstituted benzyl bromide for the formation of EDA complexes with 3-methyl-1H-indole derivatives (-9.98 to -16.59 kcal/mol, see Fig. S1-S2). For the electron-donating group, the binding energies of the EDA complexes formed with the 3-methyl-1H-indole derivatives were also all lower than unsubstituted benzyl bromide derivatives, but not numerically as large as those of the electron-withdrawing ones (-8.44 to -9.92 kcal/mol, see Fig. S1 and S3). For the bonding energies of EDA complexes formed by nitro-substituted 3-methyl-1H-indole derivatives with benzyl bromide derivatives, we found an interesting pattern. That is, in general, for the values of binding energies methoxy-substituted is greater than methyl-substituted is greater than unsubstituted (-7.75 to -16.98 kcal/mol, see Fig. S4 and S5). In summary, we found that the binding energy of EDA complexes may be related to the difference between rich and poor electrons of aromatic rings forming it, so in the next study we chose six representative EDA complexes (Scheme 2) to carry out detailed calculations and property analysis.
The structures, bonding energy and thermodynamic properties of representative EDA complexes.
The structure of the optimized EDA complex is shown in Fig. 1. Based on the analysis of the structures of the six selected EDA complexes, we found that all benzyl bromide derivatives are "face-to-face" with 3-methyl-1H-indole derivatives. Moreover, all the benzyl bromide derivatives are tilted to a certain extent during binding, and their structures are not spatially parallel to those of the 3-methyl-1H-indole derivatives. For the reasons mentioned above, we may not be able to obtain the exact distance between the two molecules. Therefore, we added a dummy atom in the plane where the indole is located in the optimized structure, and then measured the distance between the dummy atom and the specified atom on the benzyl bromide derivative and used it to estimate the distance between the planes where the two molecules are located (for details, see ESI†, Fig. S6). The results indicate that the distance between the two molecules in complex 13a-1c is the greatest
at 3.48 Å. The distance between the two molecules in complex 3b-1c is 3.16 Å, which closely aligns with single crystal data of EDA complex obtained by Melchiorre et al. Furthermore, there is only a minor difference of 0.32 Å between the maximum and minimum values, which is not significant. Therefore, we proceeded to analyze the binding energy, free energy, enthalpy and entropy values of the EDA complexes.
Table 1
The binding energies (ZPE), ΔG, ΔH and ΔS of EDA complexes 1.
| EDA complexes | ZPE | ΔG | ΔH | ΔS |
| 3b-1c | -14.70 | -0.77 | -14.43 | -0.05 |
| 13a-1c | -6.92 | 4.58 | -6.39 | -0.01 |
| 6a-1c | -9.90 | 2.28 | -9.36 | -0.02 |
| 3b-10c | -15.73 | -0.69 | -15.47 | -0.05 |
| 13a-10c | -10.57 | 1.25 | -9.97 | -0.03 |
| 6a-10c | -15.22 | -1.26 | -14.88 | -0.05 |
1 Energies in kcal/mol.
The binding energy can measure the difficulty and stability of the formation of EDA complexes. According to the data in Table 1, in general, the binding energy of 3-methyl-1H-indole with nitro (10c) and the selected benzyl bromide derivatives is greater than that of 3-methyl-1H-indole without nitro (1c). In addition, for the three 1c-complexes, the electron-withdrawing 2, 4-dinitrobenzyl bromide (3b) has the lowest bonding energy. The bonding energy of benzyl bromide without any substituents (13a) is the highest. For the 10c-complexes, we got similar result. Through the above results, we found that the bonding energy of EDA complexes formed by different substituents (electron withdrawing or electron donating) is significant. For example, the complexes formed by electron-poor benzyl bromide 3b with 3-methyl-1H-indole 1c and 3-methyl-4,6-dinitro-1H-indole 10c have energies of about − 15 kcal/mol; The complex energy of electron-rich benzyl bromide 6a and electron-poor 3-methyl-4,6-dinitro-1H-indole 10c is -15.22 kcal/mol; The binding energy of the unsubstituted benzyl bromide with 10c is lower than that with 1c. The formation of 3b-1c, 3b-10c, and 6a-10c complexes is spontaneous (ΔG values are all negative). All complexes formation processes have exothermic properties (ΔH values are negative). The ΔS values of all complexes were less than zero, which indicated that the process was an entropy decreasing and reversible process, it was also confirmed that the formation of complexes was spontaneous. These results indicate that different substituents have important effects on the stability of EDA complexes, and the electron differences between aromatic rings of EDA complexes may be one of the main reasons for their stability.
The interaction region indicator (IRI) analysis of representative EDA complexes.
Interactions between atoms are ubiquitous in chemical systems. Graphical representations of interactions are certainly useful in examining chemical problems, allowing the chemist to quickly visualize what interactions are occurring where in the system. We show the IRI analysis results of selected complexes in Fig. 2. Overall, the Π-Π interaction between the benzyl bromide derivatives and the indole derivatives is almost distributed between the two aromatic rings, and the intensity is close to the vdW interaction. The interaction area between unsubstituted benzyl bromide and indole derivatives is the smallest, and both nitro-substitution and methoxy-substitution can increase the interaction area. In addition, the interaction area of nitro-substituted indole is larger than that of unsubstituted indole.
The FMO analyses of representative EDA complexes.
As an effective method to analyze the characteristics of electron donors and acceptors, FMOs theory has been widely used. Generally accepted by researchers, the mechanism of EDA complex triggering reaction is that under the excitation of visible light, the electrons in the complex HOMO (distributed in the electron donor molecule) are excited to transfer to the LUMO orbital (distributed in the electron acceptor molecule), thus a single electron transfer (SET) process occurs. Therefore, a reasonable distribution of HOMO and LUMO as well as an energy gap in the appropriate range are necessary conditions for the creation of a functional EDA complex. Hence, it is necessary to study the distribution of HOMO and LUMO and the energy gap in EDA complexes. For the complexes formed by benzyl bromides with 1c, the distribution of their FMOs is quite similar, and HOMO always distributed on 1c and LUMO always distributed on the benzyl bromide derivatives. However, in complexes 6a-1c, a slight amount of HOMO was distributed on the 2,4-dimethoxybenzyl bromide (6a). This may be due to the fact that compound 6a carries two electron-donating substituents on the benzene ring. When we look at another set of EDA complexes, that is, the three complexes formed by the benzyl bromide derivatives with 10c, we observe that there is a large difference in the distributions of the FMOs of these complexes with respect to the complexes associated with 1c. First, in 3b-10, HOMO is distributed on 10c while LUMO is distributed on 3b. But when the two nitro groups are removed, a small amount of HOMO in 13a-10c has been distributed on 13a. Finally, when the benzyl bromide was replaced by the electron-rich 6a, the situation was reversed, with HOMO distributed mainly on 6a and to a lesser extent on 10c, while LUMO was distributed entirely on 10c. The probable reason for these phenomena is that 10c is an electron-poor aromatic ring, and thus the formation of a complex with the electron-rich benzyl bromide 6a leads to a completely opposite distribution of FMOs. Therefore, we suspect that the extent of difference between the rich and poor electrons of the two aryl rings cannot be regulated only by considering them in the pre-design of the EDA complexes. Although the Π-Π interaction between 6a-10c's is close to that of 3b-1c (which can react smoothly), the distribution of FMOs tells us that its use as a functioning EDA complex is almost impossible.
In Table 2, we list the HOMO and LUMO energies of EDA complexes and the energy gap. Among them, 3b-1c has a minimum energy gap of 4.87 eV, but 3b-10c has a large energy gap of 6.74 eV, second only to 13a-1c with a value of 6.87 eV. The energy gap of 13a-10c is 5.48 eV, which is smaller than 13a-1c. The energy gap of 6a-1c is 5.74 eV, which is 0.27 eV lower compared to that of 6a-10c. Summarizing the above data, we find that the energy gap of EDA complexes is relatively low when EDA complexes have electron deficient-rich or electron rich-deficient components.
Table 2
The EHOMO, ELUMO and Eg of EDA complexes.
| EDA Complexes | EHOMO/eV | ELUMO/eV | Eg/eV |
| 3b-1c | -6.69 | -2.09 | 4.87 |
| 13a-1c | -6.78 | 0.09 | 6.87 |
| 6a-1c | -7.56 | -1.82 | 5.74 |
| 3b-10c | -6.83 | -0.10 | 6.74 |
| 13a-10c | -7.65 | -2.18 | 5.48 |
| 6a-10c | -7.32 | -1.84 | 5.47 |
Then, we calculated the energy gap of the EDA complex in the unbound state and compared the energy gap of the EDA complex in Table 3. It was found that only 3b-1c, 13a-1c and 3b-10c showed a decrease, and the energy gap of the remaining complexes increased compared with that before the unbound state (details are shown in Table 3).
Table 3
The amount of difference in Eg before and after combining.
| EDA Complexes | Eg (unbinding)/eV | Eg (EDA)/eV | ΔEg/eV |
| 3b-1c | -4.79 | 4.87 | -0.08 |
| 13a-1c | -6.73 | 6.87 | -0.13 |
| 6a-1c | -6.93 | 5.74 | 1.19 |
| 3b-10c | -5.52 | 6.74 | -1.22 |
| 13a-10c | -7.46 | 5.48 | 1.98 |
| 6a-10c | -7.65 | 5.47 | 2.18 |
The theoretical UV-Vis spectrum of representative EDA complexes.
The redshift of the mixed solution after mixing the reactants relative to the substrate in the UV-Vis spectrum is one of the important means to detect or verify the formation of effective EDA complexes. As shown in Fig. 4a, among the three possible EDA complexes formed by benzyl bromide derivatives and 1c, only 3b-1c showed obvious redshift and has clear absorption in visible light region, while in the report of Paolo et al., 3b-1c was smoothly transformed and corresponding products were obtained. As shown in Fig. 4b, none of the three possible EDA complexes formed by the remaining benzyl bromide derivatives and 10c showed obvious redshift. Although the complexes had obvious absorption in the visible light range, we believed that this was probably due to the absorption of the 10c compound itself in the visible light region, the absorption in the visible light region of the spectrum is likely to come from 10c rather than EDA complexes. Therefore, only 3b-1c of the six EDA complexes we consider is an effective EDA complex from the theoretical UV-Vis spectrum point of view.
Spin density analysis of EDA complexes (excited triplet state).
In the reaction process that can be stimulated by light and triggered by charge transfer between EDA complexes, the electron donor molecule transfers a single electron to the electron acceptor molecule under the excitation of light, thus forming two molecular fragments with single electrons. In the above process, the main possible photophysical mechanism is that the EDA complex in the
ground state (S0) is excited to generate its corresponding excited singlet state (S1), and then through the intersystem crossing (ISC) to the excited triplet state (T1). Therefore, for EDA complexes, it is very important to study the energy of excited triplet state and the distribution of single electrons on the molecular fragment. We show energy of the T1 state (relative to the S0 state) and the spin analysis results in Fig. 5. The lowest energy of complex 3b-1c-T1 (can react smoothly) is 40.9 kcal/mol, and the energy of other complexes is 7.0 to 11.4 kcal/mol higher than it. In the distribution diagram of spin density, the spin density of EDA complexes in T1 state formed by different benzyl bromide derivatives and 1c is distributed above both benzyl bromide derivatives and 1c, but in 13a-1c-T1 and 6a-1c-T1, the bond length of C-Br bond has shown a very large extension and is close to dissociation (see Fig. S8). These observations suggest that the stability of 13a-1c-T1 and 6a-1c-T1 may be insufficient, despite the reasonable electron distribution of the excited triplet state of the complexes associated with 1c. However, for the excited triple states of the three EDA complexes associated with 10c, except 3b-10c-T1, the spin density of the remaining complexes is mainly distributed on the 10c molecule, and we can think that the excitation of electrons in 13a-10c-T1 and 6a-10c-T1 may be mainly within the 10c molecule. Therefore, we speculate that the electron-deficient donor may not be able to form effective EDA complexes.
Study on EDA complexes formed by 3-methylpyrrole and 2, 4-dinitrobenzyl bromide.
As one of the most important five-membered heterocyclic compounds, pyrrole is commonly found in many biologically active natural products and synthetic drugs.[47–53] Therefore, the development of green and mild direct modification of pyrrole method is very important and meaningful. On the basis of the above studies, we designed the EDA complex formed by 3-methylpyrrole and 2, 4-dinitrobenzyl bromide and predicted the related properties of the complex and showed them in Fig. 6. In Fig. 6a, the energy difference between the S0 and T1 of the complex 3b-methylpyrrole complex is 39.0 kcal/mol, which is smaller than that of the 3b-1c complex that can react smoothly, indicating that it is possible to excite to the T1 state under visible light. The FMO distribution and energy gap of 3b-methylpyrrole are shown in Fig. 6b. HOMO is mainly distributed on pyrrole and LUMO is mainly distributed on 3b. However, its energy gap is 0.24 eV larger than 3b-1c. We also obtain similar results to 3b-1c in IRI analysis, theoretical UV-Vis spectroscopy and spin density analysis of the T1 state of the complex 3b-methylpyrrole, (Fig. 6c-e). Hence, we speculate that this complex may be an effective EDA complex.
{kind=link}
{kind=link}