Volunteers’ information and ethical statement
Samples were collected from women volunteers, and the procedure adopted was approved by the Institutional Ethical Committee (DM/2014/101/38), Bharathidasan University. The saliva samples were collected between 8.00 to 9.00 AM from 30 healthy female volunteers (age, mean = 24, range = 19 - 30), with a prior written consent [16]. The volunteers were instructed not to consume food and/or soft drink for 10 h before the sample collection. The volunteers were also asked to brush the teeth 30 min before collection of saliva so as to prevent microbial contamination.
Sample collection and process
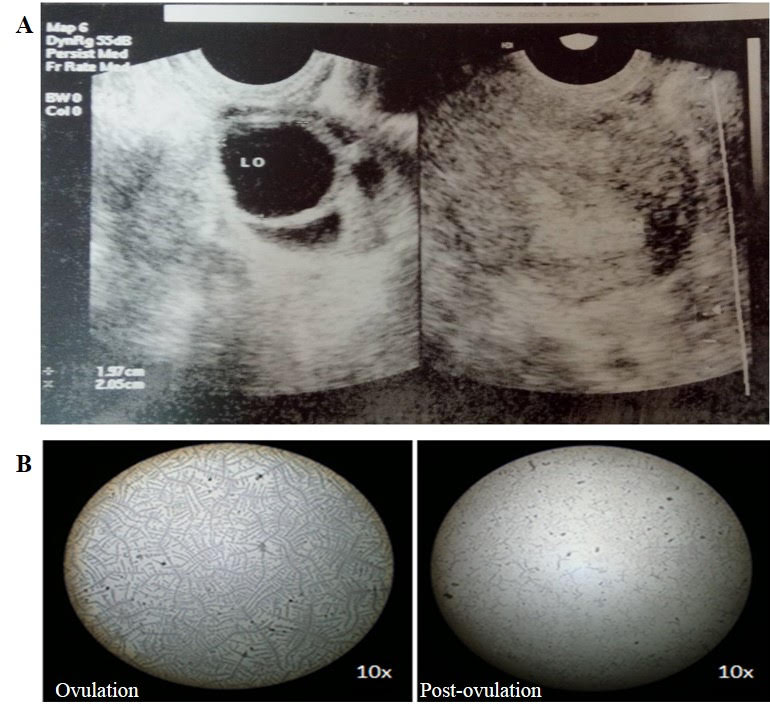
The saliva was collected by spitting method. The duration of collection of saliva was about 10 min and the saliva secretion over the first minute was discarded. The collected samples were kept in an ice box and brought to the laboratory without any time delay. The samples were centrifuged at 16000 x g for 15 min to remove insoluble materials and cells, if any. The samples were stored at -80˚C until further use. The saliva samples were assigned among three phases, viz., pre-ovulatory (day 6 to 12), ovulatory (day 13 and 14) and post-ovulatory (day 15 to 26), according to the pattern of salivary hormones and fern pattern analysis, as was done in our previous study[16].
Protein precipitation and estimation
The salivary proteins were precipitated by trichloroacetic acid (TCA)-acetone precipitation method [17]. The samples were mixed with TCA:acetone (TCA-20% W/V; Acetone-90% V/V) in 1:1 ratio and 20 mM dithiothreitol (DTT) and incubated overnight at -20 °C. After incubation, the samples were centrifuged at 5000 x g at 4 °C for 30 min. The pellets were washed twice with cold acetone and centrifuged at 5000 x g at 4 °C for 30 min. Finally, the pellets were air-dried and re-suspended in UTC (6M urea, 3M thiourea, and 8% CHAPS) buffer. The protein concentration was determined adopting the modified protocol of Bradford [18].
1D – Gel electrophoresis
To resolve the salivary proteins SDS-PAGE was carried out on 12% gel and 5-15% gradient gel (Bio-Rad) by adopting the modified method of Laemmli [19]. Standard medium-range molecular weight marker (Low-Range SDS-PAGE Standards, Bio-Rad, Hercules CA) was used as reference and it consisted of phosphorylase b (97.4 kDa), bovine serum albumin (66.2 kDa), ovalbumin (45 kDa), carbonic anhydrase (31 kDa), soybean trypsin inhibitor (21.5 kDa), and lysozyme (14.4 kDa). The salivary protein preparation from each volunteer (30 µg) was thoroughly mixed with 1x sample buffer [50 mM Tris-Cl (pH 6.8), 2% SDS, 10% glycerol, 0.1% bromophenol blue, and 100 mM β-mercaptoethanol] and kept for 1 min at 100 °C for complete denaturation of proteins, after which the sample was loaded onto the gel. A constant current of 50 V was applied for electrophoresis and the entire setup was maintained at room temperature.
Iso-electric focusing
Protein samples were mixed with an equal volume of UTC buffer (6M urea, 3M thiourea, 8% CHAPS, 100 mM DTT, and 2% IPG buffer (GE, Amersham), and incubated for 30 min in ice. The content was then diluted to the required volume using rehydration buffer (7M urea, 2M thiourea, 4% CHAPS, 0.5% ampholytes, 50 mM DTT, 1% IPG buffer (GE, Amersham), and 0.004%b blue. De-streak TM reagent (GE Healthcare) was used for better resolution. The strips were then focused in IPGphor III after 16 hr of passive rehydration. The programme used for focusing 11 cm (3-10 pH) IPG strips was as follows: 200 V-3 hrs (Step and Hold); 500 V-2 hrs (Step and Hold); 2000 V-1 hr (Gradient); 4000 V- 2 hrs (Gradient); 6000 V – 2 hrs (Gradient); 8000 V – 6 hrs (Step and Hold). The focused strips were stored at -80 ºC until further analysis.
2D – Gel electrophoresis
The frozen strips were brought to room temperature and subjected to reduction and alkylation. For reduction, the strips were incubated in SDS-equilibration buffer I (6 M urea, 50 mM Tris-Cl, 30% glycerol, 2% SDS, 0.004% bromophenol blue, and 1% DTT) for 15 min in a gel rocker. For alkylation, the strips were incubated in SDS-equilibration buffer II (6 M urea, 50 mM Tris-Cl, 30% glycerol, 2% SDS, 0.004% bromophenol blue, and 2.5% iodoacetamide) for 15 min in a gel rocker. The strips were then placed on top of 12% polyacrylamide gel (14 cm x 14 cm x 1 mm) and sealed with an overlay of 0.5% agarose solution. In the electrophoresis apparatus, the upper tank contained 2x tris-glycine buffer (0.6 % tris, 2.88% glycine, and 0.2 % SDS) and the lower tank was filled with 1x buffer. The electrophoresis conditions were 0.5 W for 45 min and 2 W for 5 to 6 hrs until the tracking dye reached the bottom of the gel plate.
Colloidal Coomassie blue staining
After electrophoresis, the gels were rinsed with distilled water and fixed with fixative solution (10% acetic acid, 40% ethanol, and 50% distilled water), following which gels were stained with colloidal Coomassie blue stain (0.02% CBB G-250, 5% ammonium sulfate, 10% ethanol, and 2% orthophosphoric acid) solution according to Dyballa and Metzger [20].
Gel analysis
Digital images of 2D-gels were acquired using ChemiDoc™ XRS imaging system (Bio-Rad) with internal calibration. The acquisition parameters were 300 dpi and epi white illumination. Gel analysis was performed by adopting PDQuest software (Bio-Rad) for spot detection, according to manufacturer’s protocol. Spot volume normalization, in the various 2-DE maps, was carried out using the relative spot volumes (% Vol). Initially, automated spot detection was performed, followed by manual editing for spot splitting and noise removal. The gels containing the largest number of protein spots for each phase were chosen as the reference gels. All other gels were matched with the reference gel by placing user landmarks on approximately 10% of major and minor protein spots, which were visualized to assist in automatic matching. Finally, all matches were checked for errors and edited manually.
HR-LC-MS/MS
The 1D protein spots were analyzed using 6550 i-Funnel QTOF-LC-MS/MS coupled with 1260 Infinity Nano pump and 1260 Cap pump along with 1260 Chip-cube (Agilent Technologies). The peptides were fractionated along with Solvent A (0.1% formic acid in milliQ water) and Solvent B (90% acetonitrile + 0.1% formic acid + 10% milliQ water). For MS measurements, we employed the positive-ion mode with the mass range of up to m/z 4000 with the resolution setting 60,000 at m/z 400. The proteins were identified by comparison with the SWISS-PROT database entries. Search parameters for MS data were, species: Homo sapiens; Protein Mass: 0–500 kDa; Protein pI: 3–10; Enzyme: trypsin; Misscleavage: 1; Mass type: monoisotopic; Charge state: MH+; precursor and product mass tolerance +/-50 and +/-100ppm, respectively; Fixed modification: carbamidomethylation of Cystine (C); and Variable modifications: oxidation of methionine (M). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository [21] with the dataset identifier PXD004511. The ovulation (File.S1) and the post-ovulation protein datasets (File.S2) were obtained and listed.
MALDI TOF/TOF analysis
2D protein spot was processed using an automated gel cutter and processor (Shimadzu, Xcise™). The gel spots were washed and destained with 50% ACN and 50 mM NH4HCO3 (Solvent 1), and subjected to in-gel digestion with 30 μL of solvent 7 (50 μLof trypsin stock solution in 4 ml of 50 mM of NH4HCO3) for 2 hr at 37 ºC. ZipTips (C18) were wetted and conditioned with 50% ACN and 0.05% TFA (Solvent 5) and 0.1% TFA (Solvent 3), respectively. Cleaved peptides bound to the C-18 resin were desalted using 0.1% TFA (Solvent 3). The peptides were then eluted and spotted with 2.5 μL of Solvent 4 (5mg/mL of CHCA in 50% ACN and 5mM of NH4CHO3) onto a 384-well MALDI plate. Finally, samples were identified using MALDI TOF/TOF (AB Sciex 4800).
Data processing
The acquired mass spectra were processed using DataExplorer® software, and the mono-isotopic peptide masses were assigned and used in the database search. The protein identification was analysed against Homo sapiens protein sequence in MASCOT database search (http//www.matrixscience.com) using SWISS-PROT database entries. Modification of cysteine by carbamidomethylation and methionine by oxidation was allowed. The precursor and product mass tolerance were set as +/-50 and +/-100 ppm, respectively. Two or more unique peptides for each protein were taken for confirmation of the protein present in the sample.
Functional annotation
The salivary proteins of the ovulatory phase were further analyzed to decipher their cellular location, molecular function and biological process by STRAP 1.5 online database (http://www.bumc.bu.edu/cardiovascularproteomics/cpctools/strap/)[22].
Molecular functional ontology
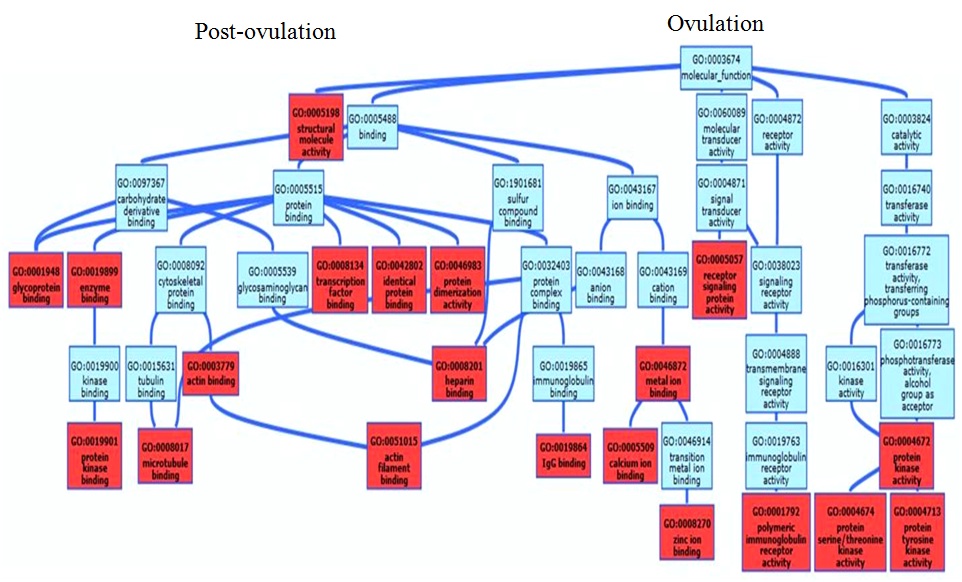
The prominent salivary proteins showing up during the ovulation and post-ovulation phases were further classified based on their cellular component, biological process, and molecular function in the UniProt database (http://www.uniprot.org/). The GO entries were used to depict the percentage of proteins through Interproscan analysis in BLAST2GO. The retrieved GO ID’s of protein entries, with their enrichment values, were used to generate a scatter plot by adopting Reduce Visualize Gene Ontology (REViGO) web server (http://revigo.irb.hr). The scatter plotting was carried out with semantic x- and y- axes corresponding to the log size value and log10 P value, respectively.
Statistical analysis
The protein concentration and band intensity values corresponding to ovulation, pre-ovulation and post-ovulation phases were represented as mean ± SD and analysed using one-way analysis of variance (ANOVA) using SPSS 16 software (SPSS Inc., Cary, NC, USA).
{kind=link}
{kind=link}