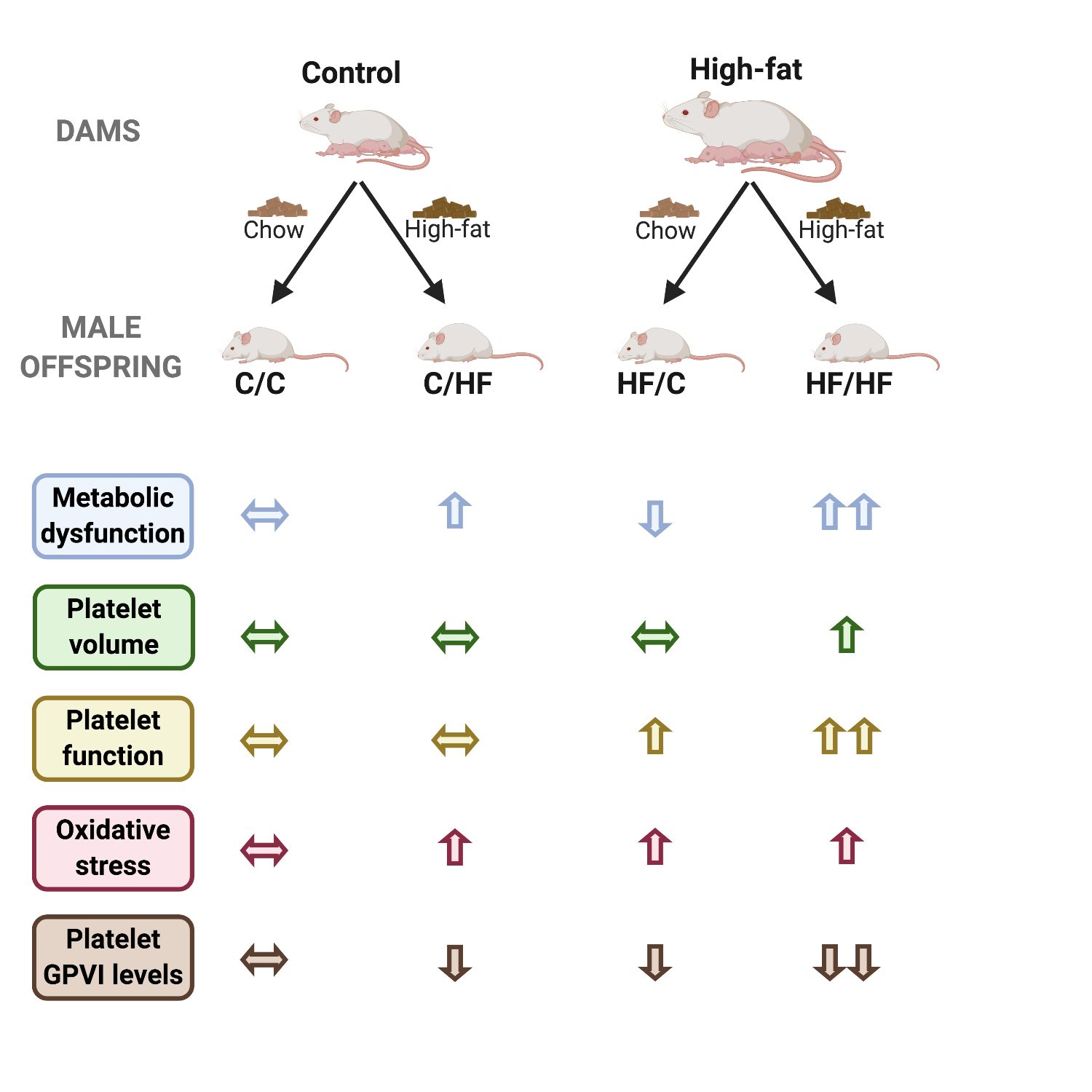
This is the first study to describe the impacts of maternal metabolic dysfunction on the platelet activity of the offspring. We show that HF animals born from HF dams presented metabolic dysfunction, with higher serum levels of triglycerides, as well as developed heavier body weight earlier than HF offspring whose mothers were lean (p < 0.01 at week 7). Maternal obesity led to increased spreading over collagen, decreased surface expression of collagen receptors and increased oxidative stress. Platelets from HF/HF animals were larger, hyper-reactive and with increased signalling, suggesting multiple mechanisms leading to increased platelet activation. Therefore, we suggest a novel ‘double-hit’ effect of maternal and offspring high-fat diet ingestion that causes platelet hyperactivation in the offspring.
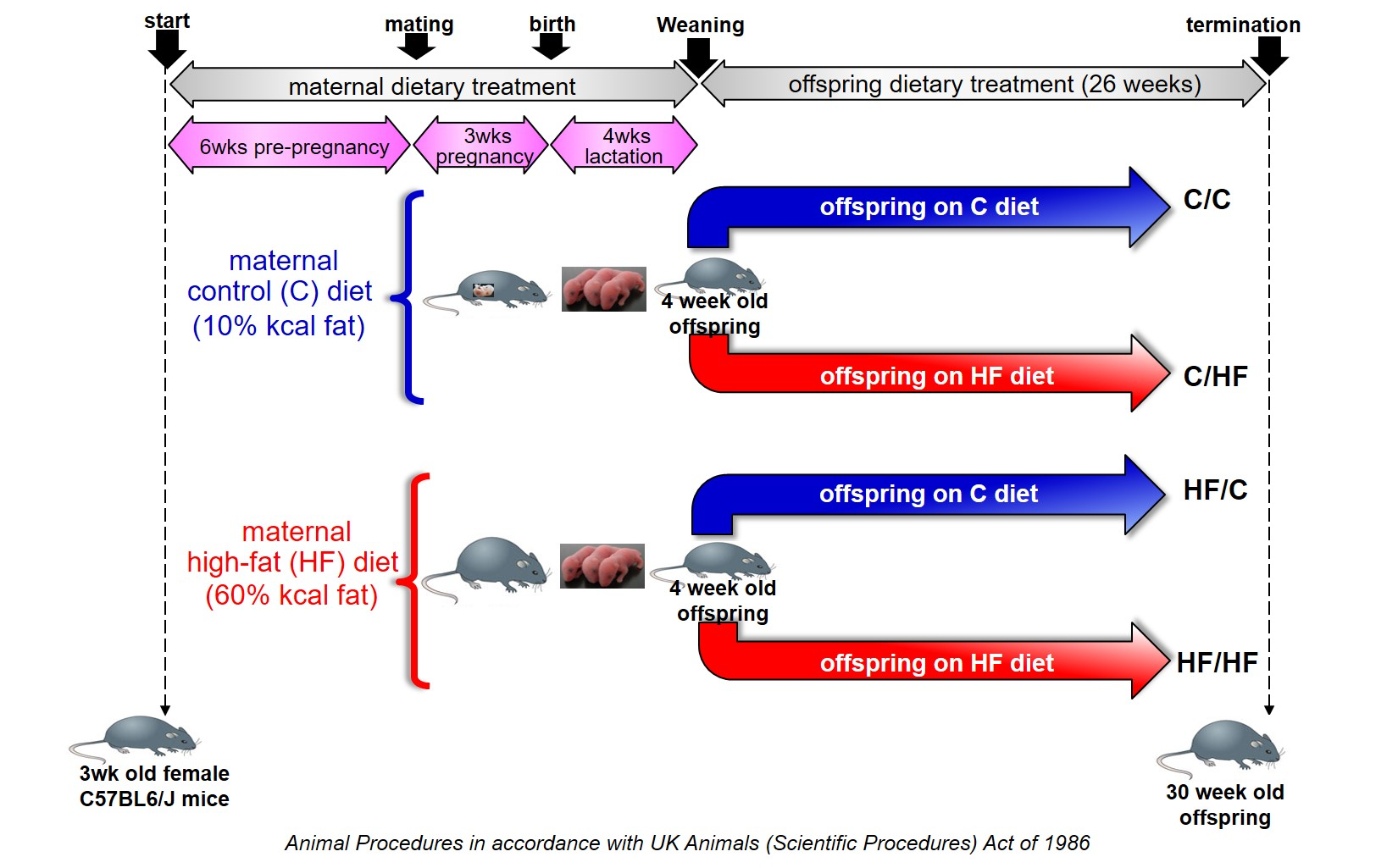
To begin with, we assessed metabolic and phenotypic parameters to characterize our model. Our data show that maternal high-fat diet decreased body weight of chow-fed offspring after 18 weeks of age, when HF/C animals began to gain less weight than C/C animals. The majority of studies assessing effects of maternal obesity on offspring have used offspring at a younger age and showed increased or unchanged body weight and adiposity in chow-fed male offspring born to obese dams [18, 19]. Similar to our data, Blackmore et al [20] showed male mice born to obese dams to have a trend towards lighter body weight and improved metabolic parameters at 12 and 8 weeks of age, respectively. They have also described an increased sympathetic activity in the heart of these mice, compared to mice born to lean dams. It is possible that maternal obesity may programme the offspring to adapt to a fat-rich environment through higher adrenergic discharge, potentially explaining the increased tendency to burn fat in HF/C offspring. This may represent an adaptive response to perceived nutritional stress such as those termed ‘predictive adaptive responses’ [21], however it is not clear why these potential adaptive changes occur at a delayed point during the life-course of the offspring. Further work is necessary to examine this potential adaptive response and indeed to confirm whether higher sympathetic activity may account for the effects on body weight.
Our results showed that if mice born to HF dams were weaned onto an obesogenic diet, these mice displayed increased body weight, adiposity and serum cholesterol levels as well as glucose intolerance, decreased RER and energy expenditure compared C/C mice. Increased body weight was comparable between HF/HF and C/HF, similar to a previous report by Loche et al [22]. The effects of maternal and offspring high-fat diet were additive on serum triglycerides levels. This is in agreement with previous reports describing a consistent [23] or a trend [18] towards increased serum levels of triglycerides in similar murine models, albeit using younger mice. Increased triglyceredaemia have been shown to aggravate other metabolic functions, such as insulin resistance (for review see [24]). Likewise, hypertriglyceredaemia was correlated with increased platelet activity both after acute intralipid injection [25] and in chronic obese patients [26]. Therefore, it is possible that the additive effect of maternal and offspring obesity on serum triglycerides may be correlated with altered platelet function and increased cardiovascular risk.
In agreement with increased serum triglycerides levels, HF/HF mice presented altered platelet size and function. These mice had larger platelets with enhanced spreading on collagen as well as activation induced by two agonists that act through distinct mechanisms. Interestingly, maternal or offspring high-fat diet ingestion had an overall effect of increasing MPV in offspring. In contrast, only maternal obesity led to increased platelet spreading over collagen, suggesting that increased platelet size does not fully explain this result. In line with this, platelets from HF/HF mice were hyperreactive to ADP and CRP, but not to thrombin. Therefore, agonist-specific effects reiterates that increased platelet function consequent of maternal high-fat diet ingestion is not fully explained by increased MPV alone.
The fact that only HF/HF mice presented increased platelet activation suggests a previously undescribed 'double-hit' effect of maternal and offspring obesity, in which both insults are needed to alter platelet function. This is in line with a previous epidemiological report describing an association between maternal obesity and premature mortality from cardiovascular events [27], to which platelets are intrinsically related [28]. Therefore, it is possible that the platelet hyperactivation herein observed in HF offspring born to HF dams can be a key pathophysiological component of the cardiovascular consequences of maternal obesity described in humans. Future research will explore epigenetic changes in platelets and megakaryocytes to better understand this phenomenon.
Platelet GPVI and integrin α2 expression was decreased due to maternal high-fat diet ingestion. We believe these alterations could be a consequence of shedding, in case of GPVI [29] or epigenetic changes in case of integrin α2. Considering that all HF groups presented increased levels of circulating ROS and that GPVI activation is both cause and consequence of increased ROS production in platelets [30], it is possible that oxidative stress interferes with GPVI expression and response to CRP. This, however, may not be the only pathway involved, given that HF/C and C/HF mice had normal platelet function, highlighting the potential for developmental programming at the epigenetic level based on the metabolic profile of the mother.
Barrachina et al have recently shown that platelets from obese individuals are hyperresponsive to CRP and that they express higher levels of GPVI when compared to non-obese individuals [31]. Using non-human primates, Arthur et al [32] demonstrated that platelets from diabetic monkeys produce more ROS and are more responsive to CRP despite having unaltered levels of GPVI. There is a lack of reports assessing platelet GPVI levels in obesity and, although not directly comparable, the abovementioned studies flag the importance of the GPVI signalling pathway to the platelet dysfunction observed metabolic diseases. We argue that this might also be true for the consequences of maternal obesity on the platelet hyperactivation seen in obese offspring.
Besides differently expressed receptor levels, platelets from HF/HF mice also showed increased PKC substrate, total tyrosine and Akt phosphorylation; an effect not seen in other groups. Although we only studied these following GPVI activation, they are all key signalling events common to a range of agonists (for review, see [33]), therefore it will be interesting to extend this work to identify whether other signalling pathways are affected or whether this is a GPVI-specific effect. Moreover, it has been shown that platelets from obese individuals are less sensitive to inhibitory molecules such as NO [34]. Our data do not support differences in NO sensitivities, suggesting that metabolic dysfunction in both maternal and offspring favours platelet dysfunction through higher sensitivity to stimulatory signals rather than lower responsiveness to inhibitory ones. Nevertheless, this should be further explored in the future with the use of other inhibitors, such as aspirin and PGI2.
We acknowledge several limitations in this study. As this is the first report on the effect of maternal obesity on platelet function, we did not exhaust all aspects of platelet function. Future research could use different approaches, such as: platelet aggregation, calcium mobilization, and thrombus formation in vitro. Likewise, epigenetic changes due to maternal obesity were not explored and could provide interesting insights on the precise mechanism of the phenotype herein observed. We recognize that data in vitro do not always translate in functional consequences in vivo and therefore encourage future reports to assess the effects of maternal obesity on thrombus formation in vivo and link animal data with human studies to support or discard our hypothesis. Finally, we believe that future studies should address sex-specific effects of maternal metabolic dysfunction on platelet function.
{kind=link}
{kind=link}