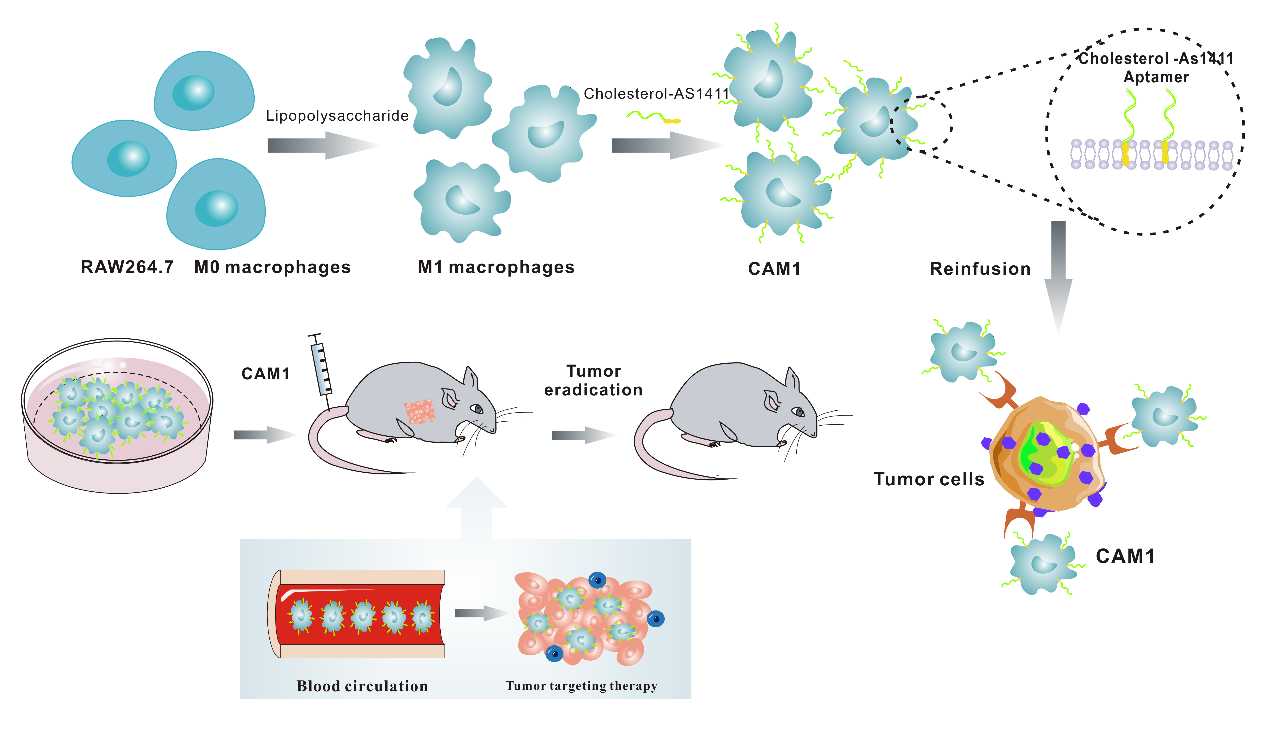
Construction of CAM1 cells
To functionalize M1 macrophages for cancer therapy, we used the lipophilicity of Chol to deliver the targeting molecule Chol-AS1411 to the surface of M1 macrophages, which were called Chol-AS1411-M1 macrophages (CAM1). Confocal microscopy analysis revealed a faint green fluorescent signal (Chol-AS1411-FAM) on the surface of RAW264.7 cells after 10-min incubation with Chol-AS1411-FAM. Furthermore, the intensity of green fluorescence on the surface of RAW264.7 cells increased with increasing incubation time. When the incubation time reached 1 h, a clear and complete circle of green fluorescence was observed on the cell membrane surface, proving that Chol-AS1411 could bind to the cell membrane surface after incubation for 1 h (Figs. 1B-C). Further determination of green fluorescence by flow cytometry indicated comparable results (Figs. 1D-E).
To confirm the stability of Chol-AS1411 in DMEM medium to ensure the sustained antitumor effect of CAM1 in vivo, M1 cell surfaces coated with Chol-AS1411-FAM inducer were incubated with DMEM for different periods, and the fluorescence signal of the CAM1 surface-modified aptamer was monitored. Confocal microscopy findings (Figs. 1F-G) indicate that the aptamer signal anchored on the CAM1 surface remained relatively stable for up to 48 h. Over time, the relative fluorescence intensity of the cell surface decreased. This study suggests that this is due to cell division during the proliferation of CAM1 cells, which also leads to a decrease in fluorescence intensity in the field of view.
In this study, RAW264.7 cells were induced into M1 macrophages by a specific concentration of Lipopolysaccharides (LPS), which can cause a strong systemic inflammatory response in vivo. Therefore, we quantified the endotoxin content of CAM1 to ensure its biosafety in vivo and in vitro. The results indicated that the concentration of endotoxin in the third wash solution of CAM1 cells was approximately 0.07 EU/mL, a value significantly below the threshold of 0.50 EU/mL established by the Chinese Pharmacopoeia (Fig. S1).
Alteration of CAM1 Activity in Vitro
The viability of CAM1 cells was evaluated using calcein-AM/PI and CCK-8 assays. The calcein-AM/PI assay results revealed no significant difference in the survival rates of M1, M1 (with AS1411), and CAM1 cells (Figs. 2A-B). CCK-8 assay was used to determine the viability of M1, M1 (with AS1411), and CAM1 cells. There was no significant reduction in the viability of the CAM1 cells (Fig. 2C). These findings indicate that CAM1 cells have good cellular activity. Moreover, the wound healing results demonstrated that the migration ability of CAM1 cells did not differ significantly from that of M0 and M1 cells in the control group (Figs. 2D-E), implying that the target molecule Chol-AS1411 did not significantly alter the macrophage migration behavior. Transwell results confirmed this finding (Figs. 2F-G).
To examine alteration of CAM1 properties, we examined the expression of CAM1 marker genes and proteins and cytokine secretion levels to determine whether CAM1 could reprogram the TME in vivo. In this study, qRT-PCR was performed to measure the expression of the CAM1 genes (Fig. 2H). The results revealed that CAM1 cells significantly expressed iNOS mRNA compared with M0 cells (vs. M0, ****P < 0.0001). This finding suggests that our modified CAM1 cells did not change the M1 polar type of macrophages. In contrast, there was a significant increase in the expression of IL-6 mRNA (vs. M0, ****P < 0.0001), IL-1β (vs. M0, ****P < 0.0001), and TNF-α mRNA (vs. M0, *P < 0.05) in M1 and CAM1 cells compared with M0 cells (Fig. 2H). ELISA results demonstrated that CAM1 cells secreted significantly more pro-inflammatory factors, such as IL-12 (vs. CAM0, ****P < 0.0001), IL-6 (vs. CAM0, ** P < 0.01), and TNF-α (vs. CAM0, *P < 0.05) compared with CAM0 cells. Concurrently, the secretion of the anti-inflammatory factor IL-10 was inhibited (vs. CAM0, ***P < 0.001) (Fig. 2I). PE anti-mouse F4/80 was then used to label mature macrophages, and FITC anti-mouse CD86 was used to label the CD86 protein on the surface of macrophages. Flow cytometry results revealed that CAM1 cells could significantly express the CD86 membrane protein compared with other groups (vs. M0, ****P < 0.0001), indicating that the modified CAM1 cells did not change the M1 cell type (Fig. 2J). Our results demonstrated that CAM1 has excellent cellular activity and the potential to reprogram the TME.
Antitumor effects of CAM1 in Vitro
AS1411 can specifically bind to NCL overexpression on the surface of tumor cells; therefore, we selected mouse breast cancer 4T1 and mouse melanoma B16 cells with positive expression of NCL for the target assay. Confocal microscopy imaging revealed that the C-AS1411-FAM (random sequence, Table S1) incubation group exhibited hardly any green fluorescence. Conversely, noticeable green fluorescence was observed in the AS1411-FAM incubation group (Figs. 3A and E), and the fluorescence intensity of the AS1411-FAM incubation group differed significantly (****P < 0.0001 vs. M0) (Figs. 3B and F), indicating that AS1411 specifically binds to 4T1 tumor cells with positive NCL expression. The fluorescence intensity on B16 tumor cells was further investigated using confocal microscopy and flow cytometry, and the results were consistent (Figs. 3C and G; D and H). Furthermore, the findings demonstrated that AS1411 had a significant affinity and was specifically bound to NCL on the surface of tumor cells. Therefore, the targeting molecule Chol-AS1411 may facilitate the specific binding of CAM1 cells to NCL-positive tumor cells.
The ability of CAM1 cells to phagocytose tumor cells was explored to demonstrate their antitumor effect in vitro. The orange-red fluorescent cell membrane probe DiI and green fluorescent cell membrane probe DiO were used to label the cell membranes of macrophages and tumor cells, respectively. After 2-h co-incubation of macrophages and tumor cells, the phagocytosis of tumor cells by macrophages was observed using laser confocal microscopy. The findings revealed that CAM1 cells had the most robust green fluorescence, indicating that they had the most potent phagocytic activity against B16 and 4T1 cells, followed by M1 cells, with M2 cells demonstrating the least phagocytic activity against tumors (Figs. 3I-J). Furthermore, CellTracker Red CMTPX and CellTracker Green CMFDA were used to label the cytoplasm of macrophages and tumor cells, respectively, to identify the phagocytosis of CAM1 cells. Flow cytometry was also used to assess the phagocytic ability of CAM1 cells. The results also revealed that CAM1 cells have a strong phagocytic ability against tumors (Fig. S2).
Antitumor effects of CAM1 in Vivo
Given that CAM1 cells successfully invaded tumors in vitro, we investigated the antitumor effect of CAM1 cells in vivo. First, we established a 4T1 subcutaneous tumor model of breast cancer in female BALB/c mice to evaluate the antitumor efficacy of CAM1 (Fig. 4A). Next, 1×105 macrophages (100 µL) were administered to each mouse following the prescribed treatment regimen via intravenous injection (i.v.) and intratumoral injection (i.t.). The Chol-C-AS1411 (random sequence) modified M1 macrophage (CCAM1) served as the negative control group.Tumor growth in the PBS (i.v.) and CCAM1 (i.v.) groups was almost unrestricted and maintained a high growth rate, indicating that CCAM1 (i.v.) could not inhibit tumor growth. M1 (i.v.) and CAM1 (i.t.) also exhibited modest inhibitory effects on tumor growth. However, tumor growth in the CAM1 (i.v.) group was significantly inhibited, and there was a significant difference (vs. PBS group, **P < 0.01), indicating that CAM1 (i.v.) produced an excellent antitumor effect (Fig. 4B). Moreover, there was no significant variation in the body weight of the mice across all groups, indicating that the engineered macrophage CAM1 had significant antitumor activity without obvious systemic toxicity (Fig. 4C).
Fluorescence imaging, which can accurately reflect the accumulation and metabolism of injected macrophages in mice, is helpful in assessing cancer conditions and cell therapy efficacy during treatment in a timely and accurate manner. The fluorescence of CY5.5 was used in this study to modify the injected engineered macrophage-targeting molecules to obtain Chol-AS1411-CY5.5 and Chol-C-AS1411-CY5.5. By co-incubation with M1 cells, Chol-AS1411-CY5.5-M1 (CAM1-CY5.5) and Chol-C-AS1411-CY5.5-M1 (CCAM1-CY5.5) were obtained. First, we evaluated the distribution of CAM1 cells in tumor-bearing mice and found that all three groups, CCAM1-CY5.5 (i.v.), CAM1-Cy5.5 (i.v.), and CAM1-Cy5.5 (i.t.), effectively emitted fluorescent signals at the tumor site (Figs. 4D-E). Second, 24 h after the injection of CAM1 cells, we assessed the distribution of CAM1-Cy5.5 (i.v.), CAM1-Cy5.5 (i.v.), and CAM1-Cy5.5 (i.t.) in the major organs (heart, liver, spleen, lung, and kidney) and tumors of tumor-bearing mice. The liver exhibited the highest concentration of fluorescence, followed by the tumor, which is consistent with the fluorescence imaging results within one week. This further indicated that the engineered macrophages could enter the tumor (Fig. 4D). The spleen, the largest immune organ, is an important site for the body to produce an immune response. The present study revealed that the volume and weight of the spleen in tumor-bearing mice were significantly higher than in normal mice, indicating that the body produces a strong immune response during the growth and treatment of 4T1 tumors, resulting in spleen enlargement.
Furthermore, a peak of the fluorescence signal at the tumor site was observed on the fourth day, implying that the injected engineered macrophages successfully infiltrated the tumor (Fig. 4F). Over time, the injected cells were gradually metabolized by the body, and the fluorescence signal became weaker. However, the fluorescence signal was still detectable one week after infusion, demonstrating that the injected engineered macrophages could effectively remain at the tumor site.
To further investigate the antitumor effect of CAM1 cells in vivo, hematoxylin-eosin (HE) staining and TdT-mediated dUTP Nick-End Labeling (TUNEL) were used to detect apoptosis of tumor tissues in tumor-bearing mice in each group after treatment. HE staining demonstrated that the CAM1 (i.v.) group had more pronounced nuclear pyknosis, reduced tumor density, and more severe tumor cell necrosis than the other groups (Fig. 4G). TUNEL staining revealed that the PBS (i.v.) and CCAM1 (i.v.) groups had almost no green fluorescence. In contrast, the other treatment groups had varying degrees of green fluorescence, with the CAM1 (i.v.) group indicating the strongest green fluorescence (Fig. 4H). The results of this experiment indicated that CAM1 cell treatment led to a large region of tumor cell necrosis and apoptosis, suggesting that CAM1 cells have potent specific killing effects on tumors, inhibited tumor cell proliferation, and promoted tumor cell apoptosis, which holds promise for developing efficient and safe tumor treatments.
{kind=link}