Preparation of acellular nerve allograft
68 SD Rats (6–8 weeks old, 220-250g) were sacrificed after anesthetized by intraperitoneal injection of an overdose of 3% sodium pentobarbital, then the bilateral sciatic nerve segments (20–30 mm) were collected. Fat and connective tissues were removed and decellularized based on previous methods with some modifications [30, 31]. Nerve segments were immersed in double-distilled water (ddH2O) for 12 h, then rinsed thrice in phosphate-buffered saline (PBS), re-immersed in ddH2O, frozen at -80℃ for 12 h. After thawing, the nerve segments were treated with 1% sodium dodecyl sulfate (SDS) (Sigma, USA) for 24 h at room temperature on a shaker at 60 rpm, then were repeatedly washed with PBS to remove SDS until the PBS wash solution became clear and no foam was generated. ANA were subsequently sealed and stored in PBS containing 100 U/mL of penicillin and streptomycin, followed by sterilization using Co60 gamma radiation for immediate use or stored at -20°C for later use.
DNA quantification
To measure the remaining DNA in decellularized nerves, we lyophilized fresh and decellularized sciatic nerves (n = 3) from rats, then weighed them and extracted the total DNA using a nucleic acid extraction analyzer. Next, the fluorescence intensity values were read on a fluorescence spectrophotometer at an excitation wavelength of 480nm and an emission wavelength of 520nm using a quantitative nucleic acid detection kit (Invitrogen, P7589). The standard curve was then plotted and the DNA content was calculated.
Scanning electron microscopy (SEM)
Normal and decellularized nerves were dehydrated using graded ethanol solutions at concentrations of 30, 50, 70, 85, 95, 100, and 100% for 10 min each. After dehydration, the nerves were dried with a CO2 extraction dryer. The coating was then photographed using SEM (6700; JEOL).
Extraction of primary ADSCs
After five 3-day-old SD rats were euthanized, the adipose tissue was collected by dissecting the inguinal region of and immediately washed with PBS. After washing off the blood, the adipose tissue was minced with microscopic scissors and then digested with 0.1% (W/V) type I collagenase (Sigma-Aldrich, USA) for 40 min in a 37°C environment. Complete Dulbecco’s Modified Eagle Medium (DMEM, Gibco, USA) supplement with 100 U/ml penicillin/streptomycin and 10% fetal bovine serum was added to terminate digestion. The tissue debris was subsequently filtered using a 100 µm nylon mesh and re-filtered using a 40 µm nylon mesh. The cell suspension was centrifuged at 1500 rpm for 5 min and the supernatant was discarded. The cell pellet was resuspended in a complete DMEM medium, seeded in 25 cm2 culture flasks, and then cultured at 37℃ in a 5% CO2 atmosphere. The solution was changed after 48 h. After reaching 80% confluence, cells were passaged and P2-P4 generations were used for subsequent analysis.
Extraction of primary BMSCs
After five 3-day-old SD rats were euthanized, the femur and tibia of rats were isolated bilaterally under aseptic conditions, washed twice in PBS, both epiphyses were clipped, and fresh bone marrow was flushed out using a 1 mL syringe aspirated with complete DMEM medium containing 100 U/ml penicillin/streptomycin and 10% fetal bovine serum (Gibco, USA). After centrifugation at 1500 rpm for 5 min, the supernatant was discarded. The cell pellet was resuspended in a complete DMEM medium, seeded in 25 cm2 culture flasks, and incubated in an incubator at 37℃ in a 5% CO2 atmosphere. The solution was changed after 24 h. After reaching 80% confluence, cells were passaged and P2-P4 generations were used for subsequent analysis.
Identification of MSCs by flow cytometry
Both MSCs were passaged to P2 generation, digested using 0.25% trypsin, a cell volume of 5 × 105 was collected from each, and cells were resuspended in phosphate PBS buffer (Corning, 21-031-CV) for subsequent analysis. FITC anti-human CD90 (BioLegend, 328107, USA), PE/Cyanine7 anti-human HLA-DR (BioLegend, 307105, USA), APC anti-human CD73 (BioLegend, 344005, USA), PE anti-CD34 (BioLegend, 343605, USA), PerCP anti-human CD45 (BioLegend, 368505, USA), and Brilliant violet421 anti-CD105 (BioLegend, 800509, USA) antibodies were incubated for 15 min in the dark. Subsequent characterization was performed with the DxFLEX flow cytometer using the FlowJo v.7,6.5 software.
Animal model
Sciatic nerve defect modeling and bridging surgery
The work has been reported in line with the ARRIVE guidelines 2.0. Female SD Rats (6–8 weeks old, 220-250g) were anesthetized by intraperitoneal injection of 3% sodium pentobarbital (30 mg/kg). Next, the right hind limb was prepared and sterilized, and the skin and musculature of the right lateral thigh were incised on a sterile operating table to expose and cut off the sciatic nerve to create a 10 mm gap. The experiment was divided into two parts. First, to investigate the in vivo response characteristics and underlying molecular mechanisms of ANA combined with BMSCs or ADSCs, eighty-four rats were randomly divided into three groups (n = 28 rats per group): ANA group, ANA + BMSC group, and ANA + ADSC group. Second, in order to explore the effectiveness of BMSCs and ADSCs on neural tissue-derived ECM materials for repairing PNI, sixty-eight rats were randomized into four groups (n = 17 rats per group): ANA group, ANA + BMSC group, ANA + ADSC group, and AUTO group. In the ANA + BMSC and ANA + ADSC groups, 25 µL ADSCs/BMSCs containing 2 × 106/ml (suspended in DMEM) was equally injected into ANA along the long axis of the 10-mm ANA using a 31G insulin syringe needle. The insulin syringe was pushed at a constant speed in 1 minute to fill the entire ANA with the suspensions of BMSCs and ADSCs, respectively, and the grafts (i.e., ANA loaded with BMSCs/ADSCs) were then sutured to the nerve ends. In the AUTO group, the dissected 10-mm sciatic nerves were turned 180° and sutured. The nerve was sutured using a 10 − 0 tension-free band sutures. Subsequently, the muscle and skin were subsequently sutured layer-by-layer using 4 − 0 sutures. All rats were sterilized with iodophor to disinfect the wounds and then housed in a 12-h light/dark cycle, with food and water ad libitum for the entire study duration.
Immunofluorescence staining
To evaluate assess the degree of decellularization and characterize the composition of the extracellular matrix, the collected ANA were fixed in 4% paraformaldehyde (PFA) solution for 24h and dehydrated using 30% sucrose solution. They were then embedded and sliced longitudinally into a thickness of 9 µm in a cryostat. The slices were washed three times with PBS and then incubated with 10% goat sealing serum for 40min. Rabbit anti-Fibronectin (1:100, Abcam, ab2413) was used as a primary antibody, and nuclear staining was performed using 4',6-diamidino-2-phenylindole (DAPI) (BOSTER, AR1176).
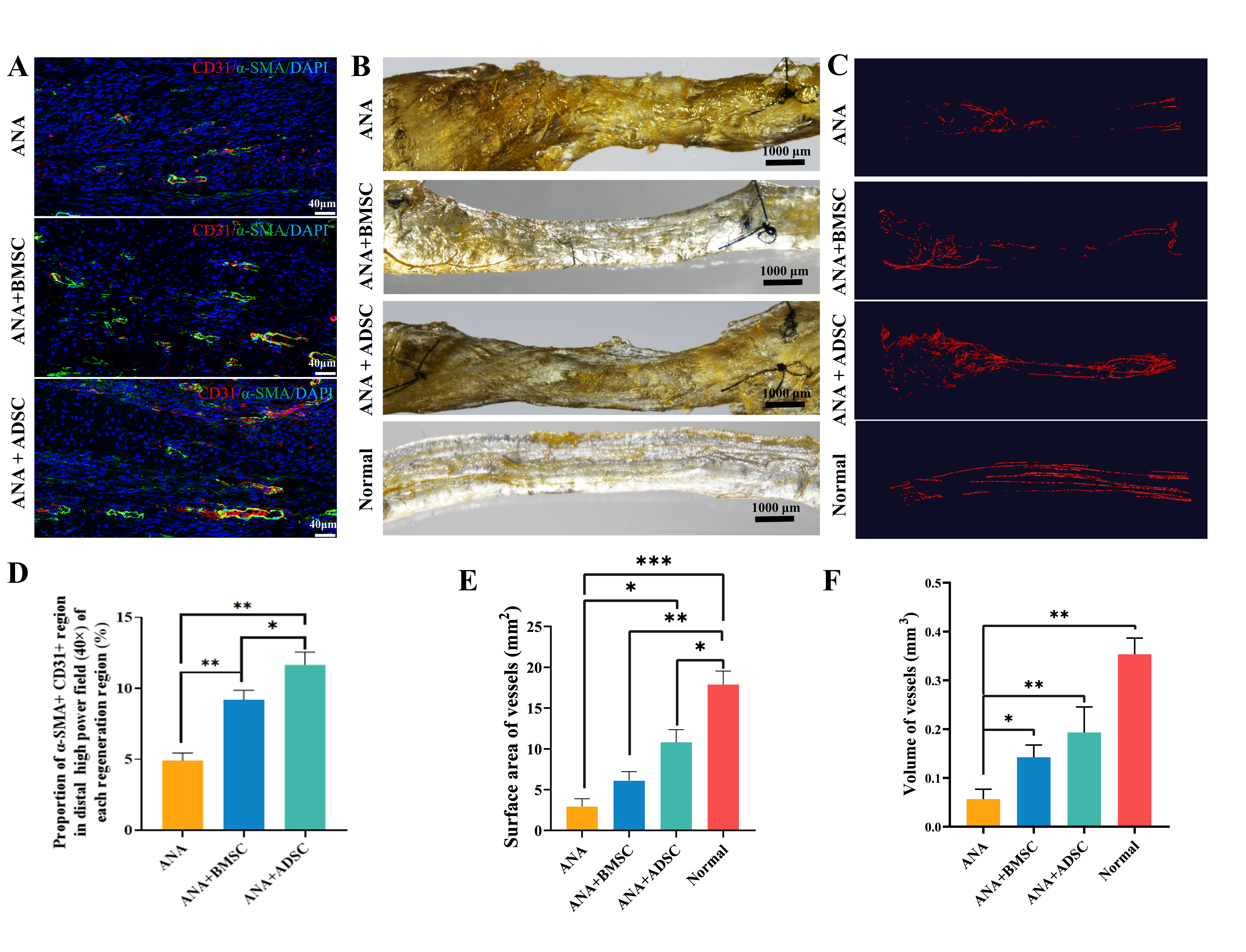
At 2 and 3 weeks postoperatively, the rats were executed by overdose of anesthesia, and the nerve grafts in each group were removed for fixation, dehydration, and slicing in the same procedure as described above. Slices were washed thrice with PBS, permeabilized with 0.5% Triton X-100 (Sigma) for 10 min, and then blocked for 40 min. Sections were incubated in a refrigerator at 4°C overnight with primary antibodies, including mouse anti-neurofilament (NF200) (1:400, Sigma, N0142), rabbit anti-S100β (1:150, Abcam, ab52642), rabbit anti-myelin protein zero (MPZ) (1:50, Abcam, ab183868), mouse anti-α-SMA (1:400, Abcam, ab7817), and rabbit anti-CD31 (1:100, Abcam, ab222783), and rewarmed at room temperature for 30 min on the following day. Then, sections were incubated with secondary antibodies, including goat anti-rabbit-Alexa Fluor®594 (1:200, Abcam, ab150080) and goat anti-mouse-Alexa Fluor®488 (1:200, Abcam, ab150117) for 2 h at room temperature. The nuclei were stained with DAPI for 10 min, and then observed and photographed under the fluorescence microscope (Nikon, Tokyo) and Pannoramic Confocal panoramic scanner (3DHISTECH).
Real-time quantitative polymerase chain reaction (RT-PCR) analysis
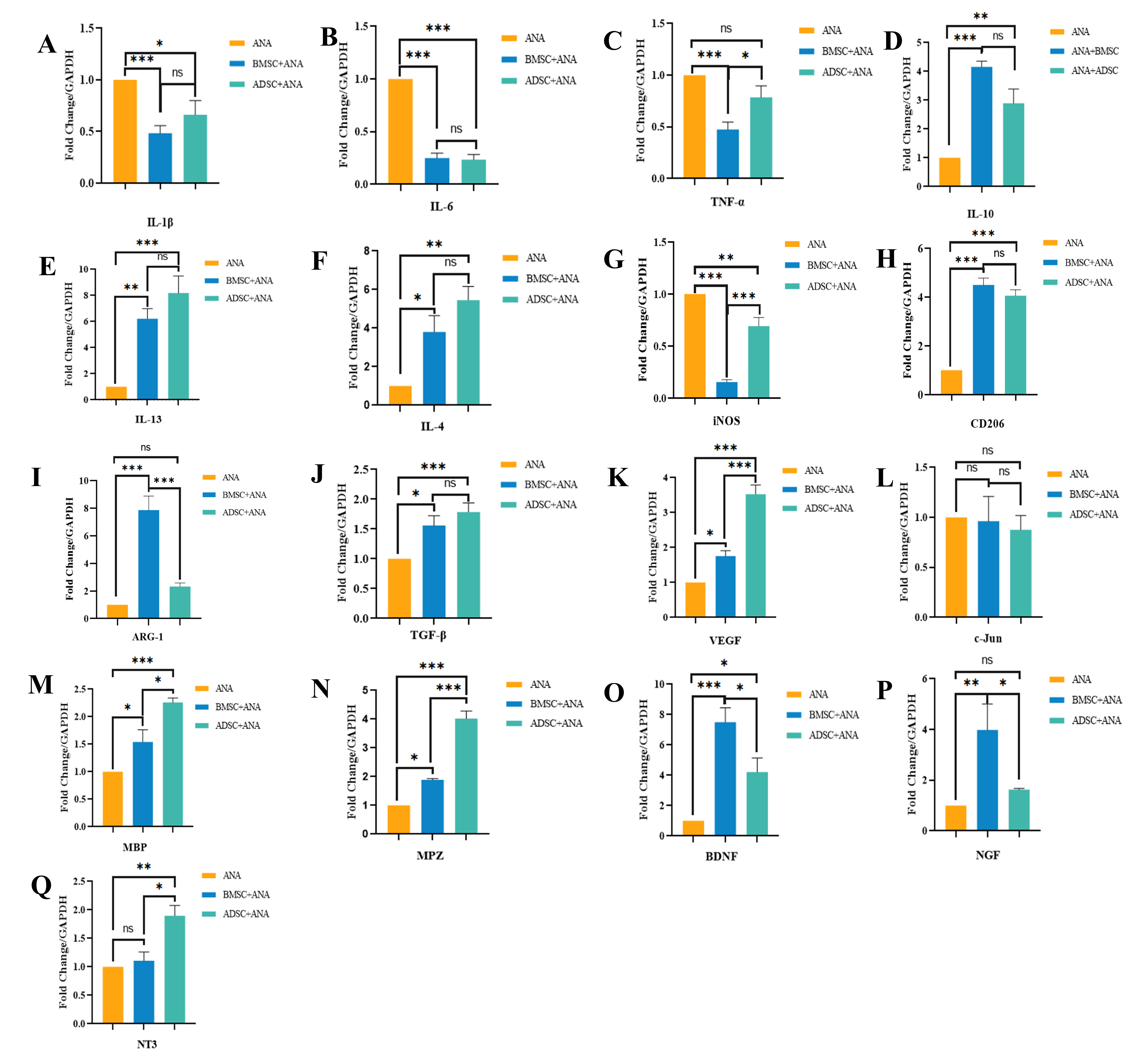
Briefly, each frozen nerve tissue was ground into a powder using liquid nitrogen, and total RNA was extracted using TRIzol (Ambion, USA). ReverTra Ace qPCR RT premix (TOYOBO, FSQ-201, Japan) was used to reverse-transcribe RNA to complementary DNA (cDNA). Subsequently, RT-PCR was performed on a StepOnePlus TM RT PCR System (Roche Diagnostics, US) using the RT2 SYBR Green qPCR Mastermix (GenStar, A303-10). The 2−ΔΔCT method was used to determine nerve regeneration-related genes (MBP, MPZ, and c-Jun), neurotrophic factor-related genes (BDNF, NGF, neurotrophin-3(NT-3)), and inflammation-related genes (IL-1β, IL-6, TNF-α, IL-10, IL-13, IL-4, inducible nitric oxide synthase (iNOS), CD206, arginase-1 (ARG-1)) at their relative expression levels. All primers were obtained from Biogenesis and their specific sequences are listed in Table 1.
Table 1
Primer sequences used in RT-quantitative polymerase chain reaction studies
| Target genes | Forward primer (5’-3’) | Reverse primer (3’-5’) |
| GAPDH | ACAGCAACAGGGTGGTGGAC | TTTGAGGGTGCAGCGAACTT |
| VEGF | GGCTCACTTCCAGAAACACG | GTGCTCTTGCAGAATCTAGTGG |
| IL-6 | TCTGCTCTGGTCTTCTGGAGTTCC | GAGTTGGATGGTCTTGGTCCTTAGC |
| IL-1β | CGACCTGCTAGTGTGTGATGTTCC | GGTGGGTGTGCCGTCTTTCATC |
| Arg-1 | CATATCTGCCAAGGACATCGT | TCCATCACTTTGCCAATTCCC |
| NGF | AAGGACGCAGCTTTCTATCC | CTATCTGTGTACGGTTCTGCC |
| iNOS | AGGCACAAGACTCTGACACCC | CGCACTTCTGTCTCTCCAAACCC |
| CD206 | TGTTTTGGCTGGGACTGACCTA | CGGGTGTAGGCTCGGGTAGTAG |
| BDNF | AAGTCTGCATTACATTCCTCGA | GTTTTCTGAAAGAGGGACAGTTTAT |
| IL-4 | CAAGGAACACCACGGAGAACGAG | CTTCAAGCACGGAGGTACATCACG |
| IL-13 | CTCGCTTGCCTTGGTGGTCTTG | GCACAGGGAAGTCTTCTGGTCTTG |
| IL-10 | CCAAGCCTTATCGGAAATGA | TTTTCACAGGGGAGAAATCG |
| TGF-β | GACCGCAACAACGCAATCTATGAC | CTGGCACTGCTTCCCGAATGTC |
| TNF-α | CGTCAGCCGATTTGCTATCT | CGGACTCCGCAAAGTCTAAG |
| C-jun | GACCTTCTACGACGATGC | CAGCGCCAGCTACTGAGGC |
| MBP | CGCATCTTGTTAATCCGTTCTAAT | GAGGGTTTGTTTCTGGAAGTTTC |
| MPZ | CATTGTGGTTTACACGGACAG | CTTGGCATAGTGGAAGATTGA |
Flow cytometry analysis
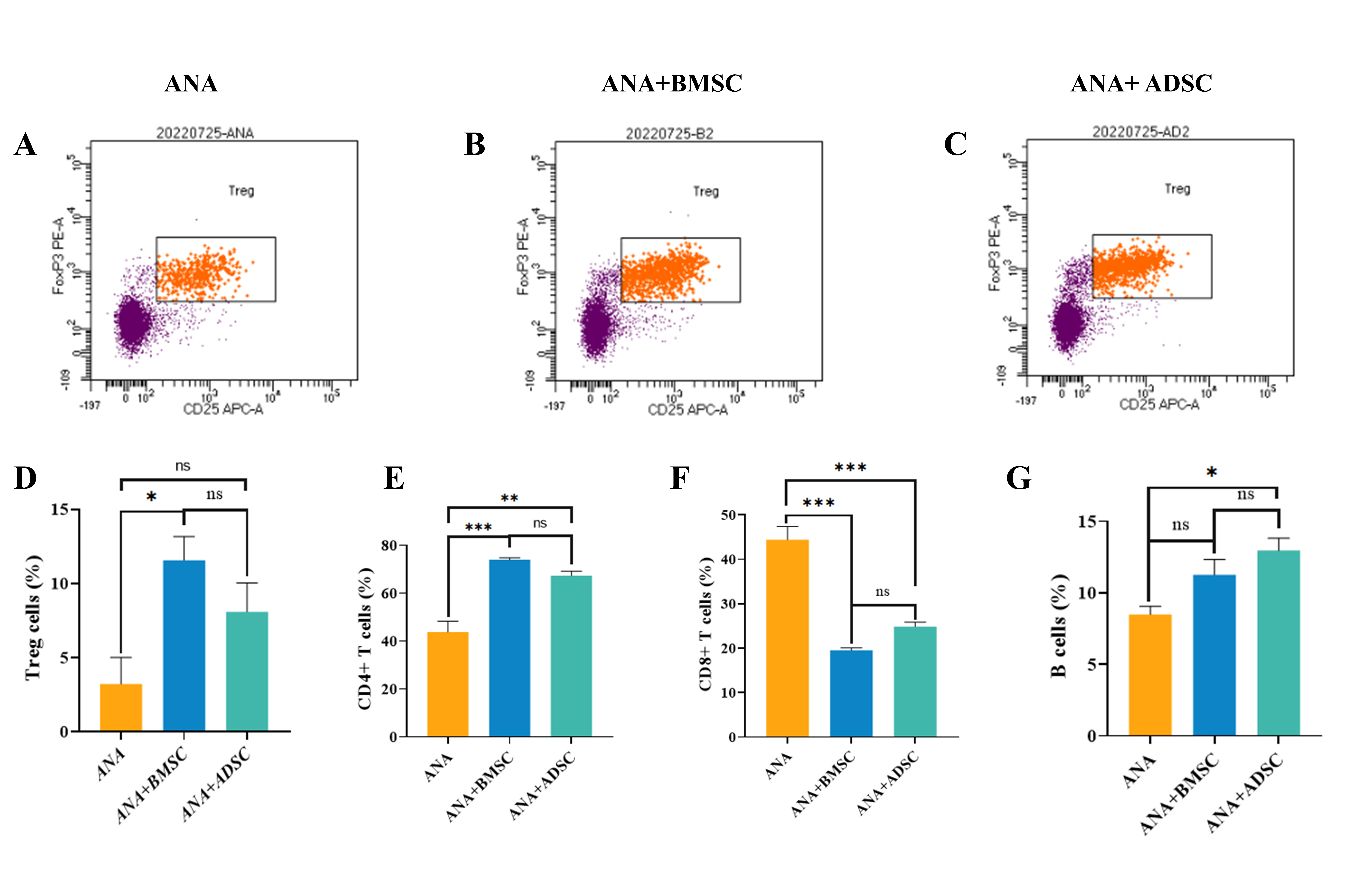
At 1 and 2 weeks postoperatively, nerve grafts in each group were obtained under aseptic conditions, sliced with scissors, and transferred to a mixture of collagenase I (0.5 mg/ml, C8490, Solarbio), collagenase II (0.5 mg/mL, C8150, Solarbio), and DNase I (25 µg/mL, D8070, Solarbio), followed by the addition of Hank’s Balanced Salt Solution (HBSS) for dissociation and incubation at 37°C for 35 min with stirring every 5 min. Immune cells were stained intracellularly and on the cell surface using antibodies after filtration. Antibodies used for flow cytometry were CD45 APC-CY7 (BioLegend, 202216, USA), CD3 APC (BioLegend, 201414, USA), CD4 FITC (BioLegend, 203305, USA), CD45RA PE-CY7 (BioLegend, 202316, USA), CD8 PercP (BioLegend, 201712, USA), CD25 APC (BioLegend, 202114, USA), FoxP3 PE (eBioscience, 17-5773-82, USA). Regulatory T cells (Tregs) were identified using the CD4 + CD25 + FoxP3 + marker. Briefly, after the addition of respective antibodies, cells were mixed and incubated for 15 min in the dark, and since FoxP3 are expressed in the nucleus, they were treated with fixation/permeabilization (eBioscience, 00-5523-00, USA) for 30 min at 4°C. All data were analyzed using the FACSCanto II flow cytometer (BD Biosciences, CA, USA) for gating analysis, and all analyses were gated to live cells first.
MicroFil vascular perfusion and micro-computed tomography (micro-CT) scanning
Rats were anesthetized and then placed in the supine position with the limbs fixed on a foam board to expose the heart. A perfusion needle was inserted into the left ventricle from the apical site to connect the perfusion pump. Then heparinized saline was perfused into the somatic circulation, followed by 2 min of perfusion using PFA to fix the vessels. Using a 20 ml syringe, the mixed MicroFil compounds were perfused through the perfusion needle into all vessels of the rats. After dehydrating the tissue with gradient alcohol and hyalinizing the tissue with wintergreen oil, the nerves were photographed in a gross view using a stereomicroscope. All nerves were scanned using micro-CT (PerkinElmer, USA) at 70 kv with 114 µA current and 20 µm resolution. Each nerve was scanned for approximately 20 min and the surface area and volume of the vessels were calculated using Analyze 12.0 (AnalyzeDirect, USA).
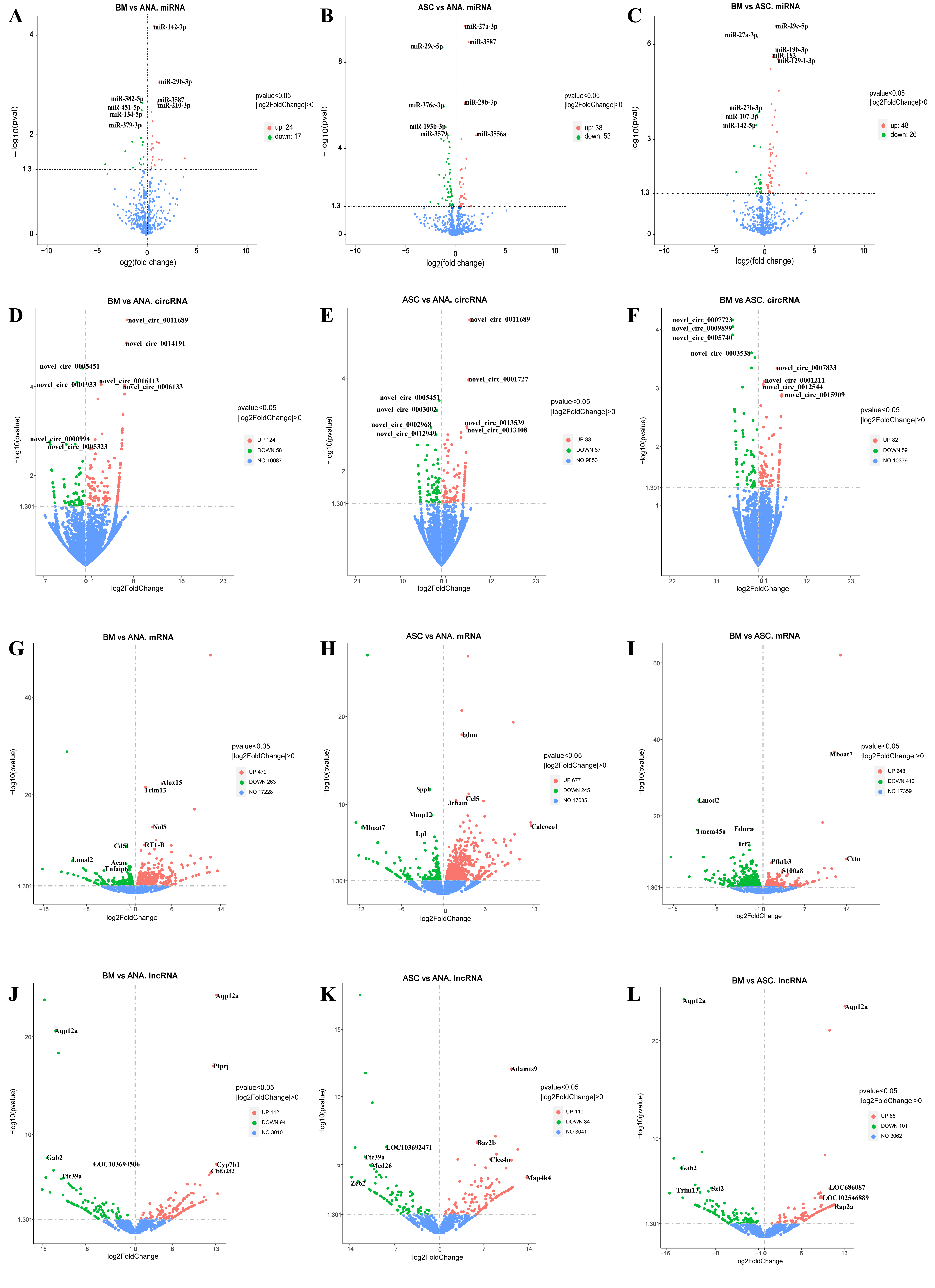
Whole transcriptome RNA Sequencing
All experiments were performed in triplicate. All the technical operations and analyses were performed by Beijing Novozymes Company (Beijing, China). Briefly, RNA was extracted and purified from each sample in each group using Trizol, and agarose gel electrophoresis was utilized to analyze the integrity of RNA and the presence of DNA contamination. Strand-specific libraries were constructed by removing ribosomal RNA (circular RNA (circRNA) library construction and addition of linear RNA removal process), and long noncoding RNAs (lncRNAs), microRNAs (miRNAs), and circRNAs were obtained. Qubit was used for preliminary quantification, and libraries were diluted to 1 ng/µl, and the insert size of the libraries was detected using an Agilent 2100 bioanalyzer. The distribution of the insert size was around 250–300 bp, which was in line with the expectation. The effective concentration of the library was accurately quantified by qPCR to ensure the library quality. In the same principle as mRNA quantification, Known_lncRNAs and predicted novel_lncRNAs, as well as the known and novel circRNAs in each sample, were identified for expression and normalization.
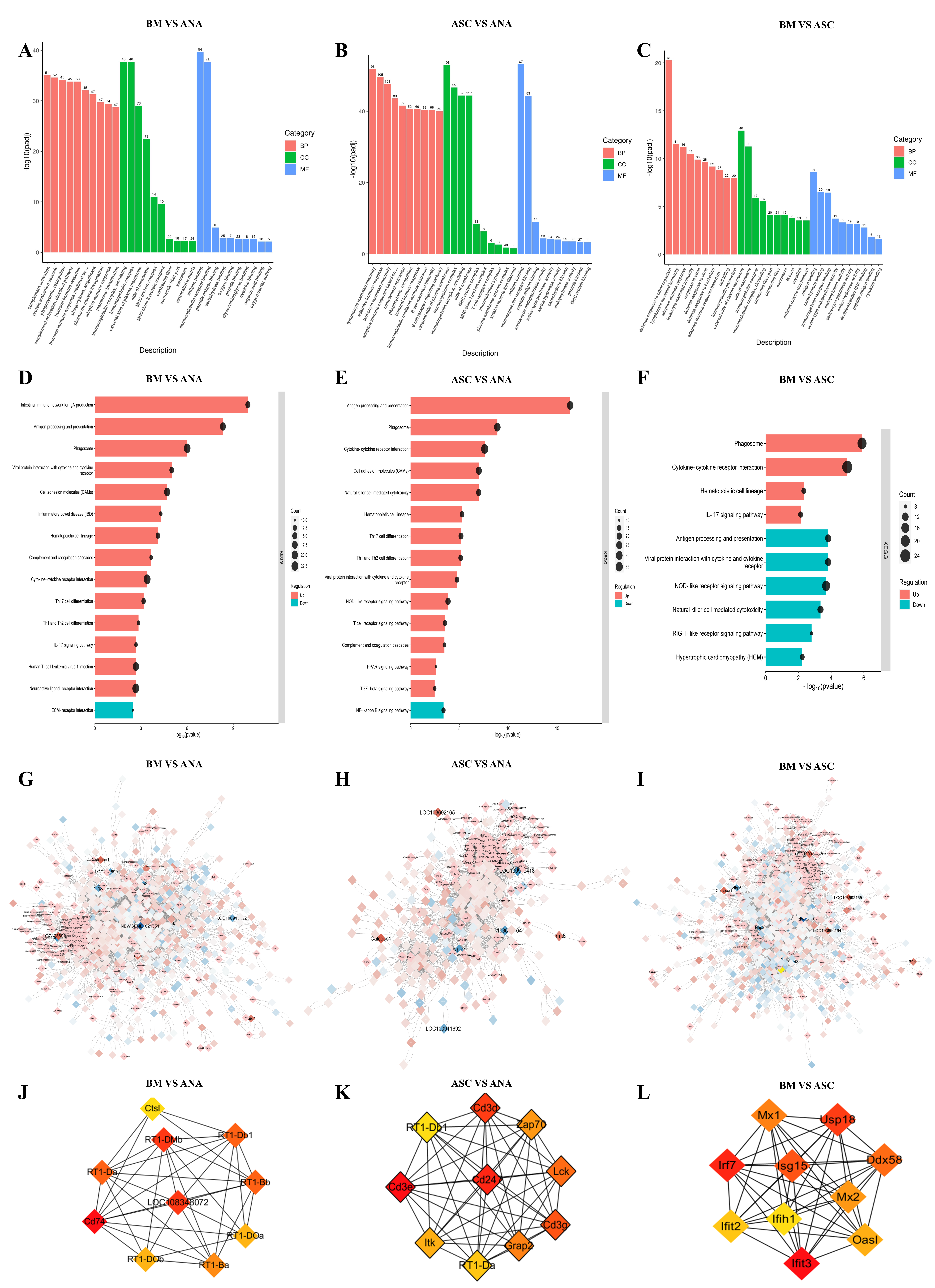
We adopted whole-transcriptome RNA-sequencing to identify differentially expressed (DE) mRNAs, miRNAs, lncRNAs, and circRNAs. Then Gene Ontology (GO), which includes cellular components (CCs), molecular functions (MFs), and biological processes (BPs), and The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DE-mRNAs was performed based on the correspondence between lncRNAs and their source genes. The criteria for differential gene screening were padj < 0.05 and | log2(fold change) | > 1.
Construction of protein-protein interaction (PPI) network and identification of hub genes
The corresponding PPI networks were constructed and plotted using the DE-mRNA in the STRING system (https://cn.string-db.org/) and Cytoscape software version 3.8.0 (San Diego, CA, USA). These isolated genes that do not interact with other proteins would be removed, and then we get a PPI network map of the target genes. And the top ten hub genes in each group were screened out using the filter parameters of the CytoHubba plugin. In the graph, the colors represent the number of correlations of the expressed modules, with the redder colors indicating more core roles. The pattern of edges denotes the regulatory relationships.
Motor function recovery assessment
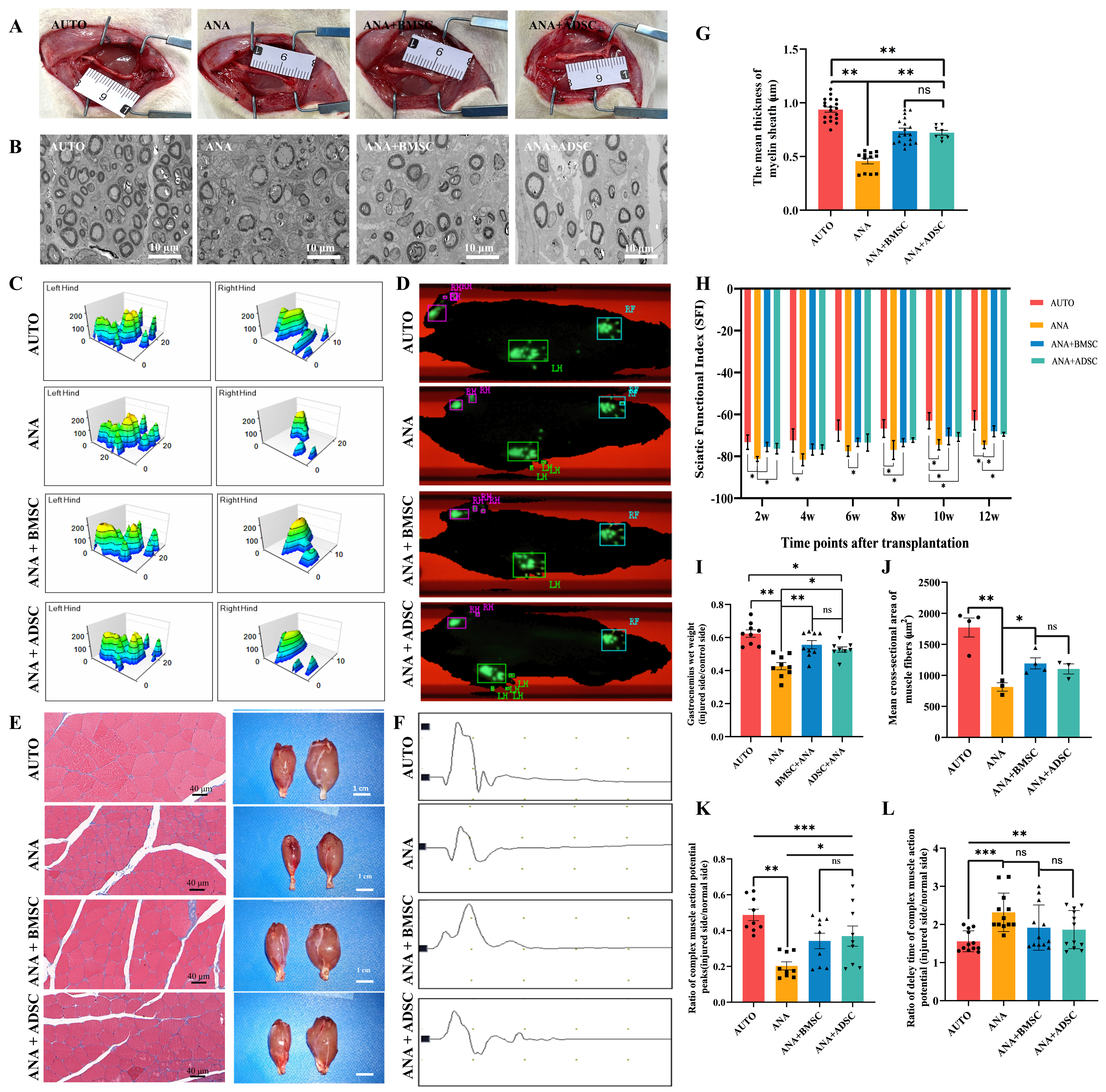
Paw prints were collected and Gait of walking rats including position, surface area, and pressure of each paw in each group was recorded and analyzed, at 2, 4, 6, 8, 10, and 12 weeks postoperatively using CatWalk XT version 10.6 (Noldus). Based on the paw prints, the system identified the paw print length (PL), toe width (TS), and intermediate toe width (ITS) of the normal side (N) and the injured side (E), and calculated the sciatic function index (SFI), which was calculated as follows: SFI = -38.3 × (EPL-NPL)/NPL + 109.5 × (ETS-NTS)/NTS + 13.3 × (EITS-NITS)/NITS-8.8. Generally, the SFI ranges from 0 for healthy nerve function to -100 for complete dysfunction.
Electrophysiological testing
Electrophysiological testing was performed at 12 weeks postoperatively. Briefly, sciatic nerves were exposed bilaterally after the rats were satisfactorily anesthetized, and electrical stimulation (intensity 3 mA) was performed sequentially at the proximal and distal ends of the nerve trunks, with the stimulating electrodes of the Synergy electromyograph (Medtronic Skovlunde) placed at the proximal end of the grafts and the parallel recording electrodes placed at the gastrocnemius muscle of the ipsilateral side, and the compound electromyographic waveforms were recorded in a single stimulation. The normal side was subjected to the same operation. The compound muscle action potential (CMAP) wave amplitude and latency ratio of the injured/normal side muscles for each group were calculated to evaluate the recovery of nerve conduction.
Transmission electron microscopy (TEM)
At 12 weeks postoperatively, after the electrophysiological tests, we sampled the nerve tissues 5 mm distal to the grafted segments in each group in order to evaluate the regeneration of nerve fibers and myelin sheaths. Briefly, the obtained neural tissues were immersed in 4% glutaraldehyde solution for 24h and in 1% osmium tetroxide for 2h at 4°C, followed by gradient dehydration in ethanol solution. Ultrathin sections of 70 nm thickness were performed using an ultrathin sectioning machine. Sections were staining using 3% lead citrate-uranyl acetate staining solution, then observed and photographed using a TEM. We counted the myelin thickness in the electron microscopic images of each group and calculated the average myelin thickness. We used Image pro plus 6.0 software to count the myelin thickness in TEM images of each group and calculate the average myelin thickness.
Histomorphometric examination of the gastrocnemius muscle
After electrophysiological tests were completed at 12 weeks postoperatively, bilateral gastrocnemius muscles were taken, weighed, and photographed, and the muscle wet-weight ratio was calculated. Subsequently, the muscles of each group were fixed in 4% PFA, dehydrated, embedded, subjected to transverse paraffin sectioning, deparaffinized, and stained using the Masson’s trichrome staining kit, according to the manufacturer’s instructions, and then observed under the microscope and photographed. Then we measured the average cross-sectional area of muscle fibers in each group using Image pro plus 6.0 software.
Image analysis
Axonal regeneration lengths (the mean length of NF200-positive staining) were measured using dedicated case viewer software. Subsequently, 3–5 random 40x microscope fields of view were captured from the fluorescence sections stained for MPZ, CD31, and α-SMA. These fields were used to quantify the extent of regeneration: average fluorescence intensity of regenerated myelin sheath (MPZ-positive staining) and average percentage of the area of functionalized blood vessels (CD31 + α-SMA + area/total area) in each field. And the mean thickness of myelin sheath was measured from 3–5 areas randomly selected from each TEM image (10x). Image analysis software (Image Pro Plus 6.0) was employed to perform cell counting and quantify early vascular regeneration, axonal outgrowth, and myelin regeneration.
Statistical analysis
Both plotting and statistical analysis were performed using GraphPad Prism 8.0 software. Comparisons among three or more groups were performed using one-/two-way ANOVA and Student’s t-test was used for comparison between two groups. All data are expressed as mean ± standard error. Differences between groups were considered statistically significant when *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}