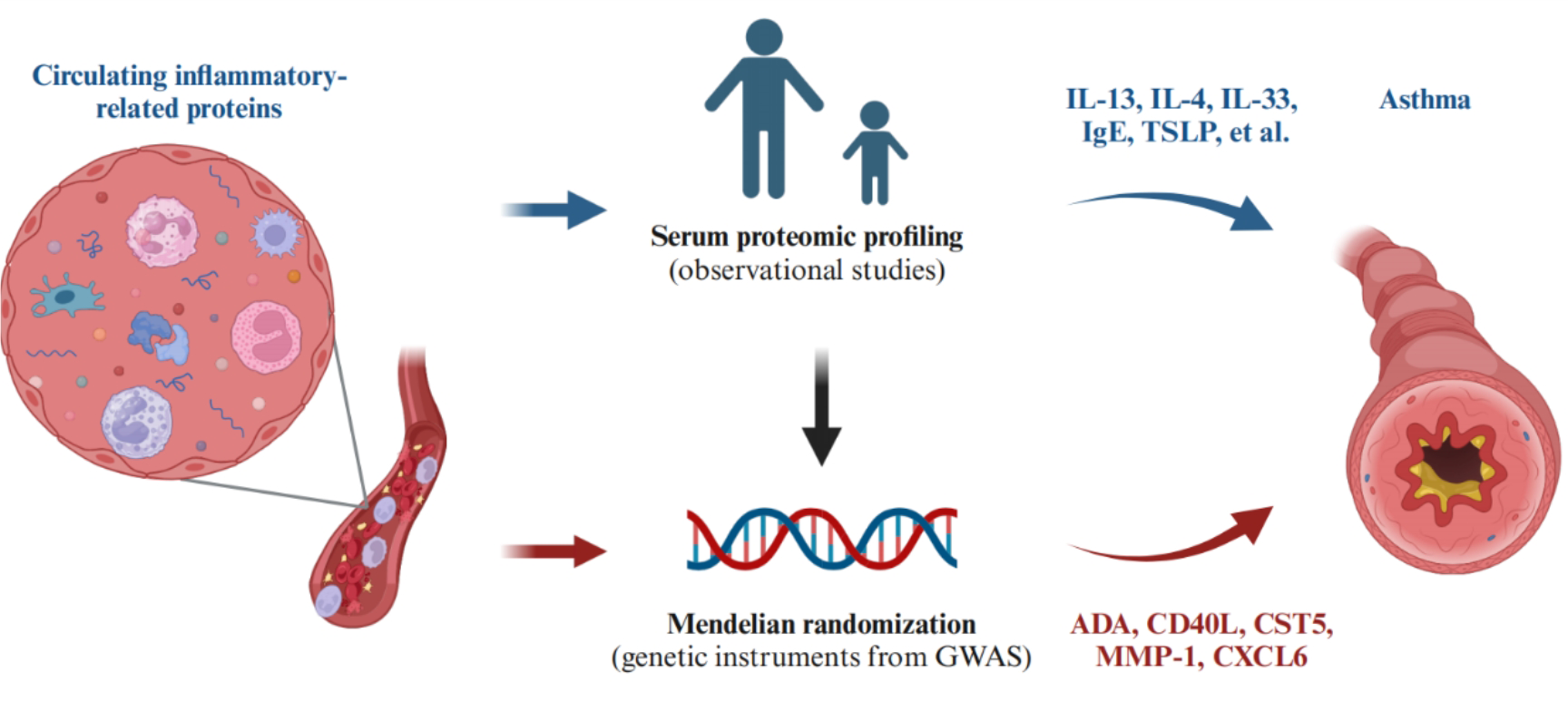
In this study, we conducted a thorough investigation into the causal associations between 91 circulating inflammatory-related proteins and asthma. We identified five protein markers (ADA, CD40L, CST5, MMP-1, and CXCL6), with CXCL6 being specific to COA, while four were shared among subsets. CD40L, CST5, and MMP-1 were supported by colocalization analyses. Bidirectional MR and Steiger filtering indicated that none of the identified proteins showed reverse causality.
Pathway enrichment analysis revealed that the potential identified targets were mostly involved in inflammatory pathways like IL-17, NF-κB, and TNF signaling pathway, all associated with asthma development. A study suggested that IL-17 promotes the development of allergic asthma by enhancing airway dendritic cell (DC) activation, migration, and function[31]. Inhibition of the NF-κB signaling pathway could alleviate airway inflammation in asthma[32]. Natalie et al.[33] indicated that the elevation TNF levels are linked to clinical phenotypes of asthma, including neutrophilic and severe asthma. In addition, the potential targets mainly participated in immune system dysregulation, such as primary immunodeficiency and RA. This suggests that the identified proteins may play a role in the systemic immune response in asthma.
CD40L, a membrane-bound protein of the TNF superfamily, is crucial for mediating the interaction between antigen-presenting cells (APCs) and lymphocytes[34]. Its engagement with CD40 plays a protective role in asthma by regulating the balance between Th1 and Th2 cells[35]. Notably, CD40L levels are significantly decreased in patients with allergic asthma[36], which consistent with our results. Our PPI analysis revealed that CD40L interacts with numerous cytokines targeted by monoclonal antibodies used in asthma treatment. Currently, CD40L is undergoing clinical trials for type 1 diabetes mellitus and kidney transplant. In addition, it was externally validated in two plasma proteins GWAS datasets. Therefore, it holds promise as a new druggable target for asthma. However, some studies have produced paradoxical results, suggesting that platelet depletion and CD40L depletion could attenuate asthma progression by inhibiting IL-4, IL-13, and IgE production, as well as leukocyte infiltration[37]. This controversy may stem from the diverse functions of CD40L across different cell types or phases of the immune response. Further investigation is needed to elucidate the detailed mechanism and biological significance of the CD40/CD40L interaction in asthma.
The matrix metalloproteinases (MMPs) function in degrading extracellular matrix (ECM) components, including collagen, laminin, fibronectin, and others. MMP-1 is involved in degrading various types of collagen like I, II, III, VI, and X[38], and is recognized as contributing to airway inflammation, tissue remodeling, and asthma exacerbation[39]. Serum MMP-1 levels are elevated in chronic asthma and may distinguish between moderate and severe asthma[40]. MMP-1 overexpression has been observed in airway epithelial cells, inflammatory cells, and airway smooth muscle cells in asthma[41], particularly in fatal asthma cases[42]. Our results support these findings. MMP-1 is currently undergoing clinical trials for lung cancer and breast cancer, among others. Moreover, we have identified three potential drugs targeting MMP-1, providing a therapeutic approach for asthma. Molecular docking has reinforced the reliability of this association through molecular binding. Furthermore, external validation was conducted in independent cohorts.
Adenosine deaminase (ADA) deficiency leads to the accumulation of toxic purine degradation by-products, most potently affecting lymphocytes, resulting in ADA-deficient severe combined immunodeficiency[43], which includes asthma[44]. Adenosine induces Ca2+ oscillations and constriction in airway smooth muscle cells, ultimately leading to calcium-induced release. Epithelial injury and airway hyperresponsiveness are hallmarks of asthma. Removal of adenosine by ADA mitigates local epithelial injury and airway contraction, thereby alleviating asthma[45]. ADA is in clinical trials for leukemia, lymphoma, and kidney cancer. Additionlly, we conducted molecular docking to further validate the interaction strength between ADA and two small molecule drugs. Our results were also confirmed by eQTL analysis in lung tissue, providing further evidence for the causality between ADA and asthma. The findings suggest that ADA might be a therapeutic target.
Additional druggable targets identified were CST5 and CXCL6. CST5 (Cystatin D) is a salivary cysteine protease inhibitor that blocks coronavirus replication at its physiologic concentration. Notably, CST5 levels were lower in COVID-19 patients compared to non-COVID-19 individuals[46]. We discovered that circulating ADA protein reduces the risk of asthma, which was validated at the genetic level in lung tissue. However, previous studies on CST5 in asthma are limited. Both CST3 and CST5 belong to the cystatin superfamily of protease inhibitors, with CST3 levels found to be higher in asthma compared to controls[47]. Further research is needed to explore the expression and function of CSTA in asthma.
CXCL6, a member of the CXC chemokine family, serves as a chemotactic agent for neutrophil granulocytes. Verhoeckx[48] proposed that upregulation of CXCL6 could exacerbate the inflammatory progression of asthma. In addition, our PPI analysis revealed that CXCL6 interacts with IL-6 and TNF-α, therapeutic targets of Tocilizumab and Golimumab, respectively, both licensed medications for asthma. Thus, CXCL6 may represent a promising new target for asthma treatment.
Several limitations should be considered when interpreting our findings. First, our analysis was restricted to individuals of European ancestry, potentially limiting the generalizability of our results to other ancestries. Further stuides involving non-European populations are necessary to broaden the discovery of genetic determinants of asthma. Second, all identified proteins had three or fewer SNPs, limiting alternative MR appproach, heterogeneity tests, and pleiotropy tests. However, the SNPs we selected were strong instruments with F statistics exceeding 30, indicating minimal weak instrumental variable bias. Third, colocalization analysis did not support ADA, CXCL6 and asthma sharing the same causal SNPs. Nonetheless, this does not invalidate the findings as colocalization methodologies typically exhibit a high false negative rate (around 60%). Finally, non-linear effects, time-dependent effects, or inflammation-environment interactions may exist between some proteins and asthma. Considering the possibility that a protein may influence asthma risk at extremely low or high levels is intriguing. However, detecting such effects in practical clinical settings could be challenging.
{kind=link}