2.1 Peptide synthesis
The designed peptides were chemically synthesized using Solid Phase Peptide Synthesis (SPPS) method using rink amide resin and Fmoc-chemistry. The resin is weighed according to the scale of synthesis and peptides are synthesized sequentially from C-terminus to N-terminus. Fmoc group is cleaved using 20% piperidine solution in DMF for 30 minutes. pH of the solution is neutralized by washing the resin 8–10 times using DMF and Fmoc-protected amino acids along with activators HoBT, HBTU and DIPEA added in 6 times excess. Attachment of amino acid is performed twice for a period of 1 hour and second time for 30 minutes. After attachment and washing with DMF, the resin is again subjected to treatment with 20% piperidine and the next amino acids are added sequentially in a similar manner. Once all the amino acids are attached, we proceed for the attachment of the fluorophore 5(6)-carboxyfluorescein and the drug methotrexate. Once the synthesis is complete, the peptides are deprotected and cleaved from the resin using a mixture of 1,2-ethanedithiol, m-cresol, thioanisole, and trifluoroacetic acid. The peptides are filtered using glass wool and precipitated and washed several times with ice-cold diethyl ether.
2.2 Purification and characterization of synthesized peptides
Synthesized peptides were purified by reverse-phase High Performance Liquid Chromatography (RP-HPLC) using a C-18 semi-preparative column (250 × 10 mm, particle size = 10 µm, pore size = 120 Å). Gradient using 10% acetonitrile in ultrapure water with 0.1% trifluoroacetic acid as solvent A and 100% acetonitrile with 0.1% trifluoroacetic acid as solvent B. HPLC peaks were collected and analysed using mass spectrometry. MALDI-TOF (Bruker, Autoflex Speed) machine in reflectron mode is used to determine the molecular mass of the peptides using α-cyano-4-hydroxycinnamic acid (HCCA) matrix.
2.3 Characterization by circular dichroism (CD) spectroscopy
Purified peptides at 50 µM concentration were prepared by dissolving in ultrapure water. CD spectra of the peptides was recorded using Jasco J-1500 spectropolarimeter by scanning between 190 nm and 280 nm using a quartz cuvette of 1 mm pathlength. Each spectrum has an average of 8 scans and the data was converted to mean residual ellipticity as described [31].
2.4 Electrostatic profiling of the peptides
Delphi software was used to calculate the Poisson-Boltzmann electrostatic potentials (PB-EP) using the Finite Difference Poisson-Boltzmann equation [32]. The output file was saved into phi format. The PB-EP was mapped on the molecular surface of peptides, analysed and rendered using Pymol. The potentials are in the units of kT per unit charge (e) where k is the Boltzmann constant and T is the absolute temperature. Each peptide has a unique electrostatic fingerprint that correlates with its functional properties.
2.5 Molecular Dynamics Simulation studies
GROMACS program suite was used to perform Molecular Dynamics Simulations [33]. GROMOS96 forcefield with 53a6 parameter set together with Berger lipids forcefield were used for simulations [33, 34]. The 1-palmitoyl-2-oleoylglycero-3-phosphoglycerol (POPG) lipid bilayer structure was obtained from the MemBuilder server [35]. The POPG forcefield parameter from former research work by Kukol et al. was employed [36]. The lipid bilayer was positioned in the XY plane, while its thickness was determined in the Z plane. Four peptide molecules were positioned on top of the lipid bilayer in the Z plane. The system was then packed with water molecules, followed by a 200 ns production run [11], [12], [37].
Briefly, a 128-membered POPG lipid bilayer was generated. Four peptide molecules were positioned close to one of the outer edge of the bilayer. The system was subsequently solvated with water and its energy was minimized. Water was used as a solvent (simple point-charge model, SPC). Pressure coupling was achieved using the Parrinello-Rahman barostat, and the isothermal compressibility was set at 4.5 × 10− 5 bar − 1. Energy minimization (steepest descent) until a 1000 KJ 1000 KJ mol− 1 nm− 1 tolerance was the next step. Subsequently, it was equilibrated for one nanosecond (ns) in NPT (conserved Number of particles, Pressure, and Temperature) conditions.
A 200 ns production run followed equilibration with four peptides on a 128-membered POPG bilayer. Water (SPC) was used as the solvent. The Root Mean Square Deviation and radius of gyration were determined for the conformations of the peptides using the g_rms and g_gyrate tools of the GROMACS package. Conformational analysis and free energy calculations were carried out based on the output of g_cluster program of the GROMACS package.
2.6 Cell culture
Human cancer cell lines MDA-MB-231 (breast cancer) and A549 (lung cancer) were procured from National Cancer Institute (NCI), USA. HeLa (cervical cancer) was obtained from Central cell line repository of RGCB. These cells were cultured in Dulbecco’s Modified Eagle Media (DMEM) with the addition of 10% fetal bovine serum (FBS) and 1% antibiotic mixture (10 mg/mL streptomycin, 10,000 units/mL penicillin and 25 µg/mL Amphotericin B) [31]. Non-tumorigenic breast epithelial cell line MCF-10A was cultured in Mammary Epithelial Basal Medium supplemented with 10% fetal bovine serum, bovine pituitary extract (BPE), hydrocortisone, hEGF, insulin and gentamicin/amphotericin-B. Lifetime imaging facility of RGCB is acknowledged for FRET-based Caspase sensor cells MDA-MB-231 SCAT3 NLS. All the cells were grown in a humidified incubator at 37 ºC with 5% CO2.
2.7 Comparative uptake of peptides by flow cytometry
70,000 cells of MDA-MB-231, A549, HeLa and MCF-10A were seeded in 24-well plates and placed in an incubator at 37 ºC overnight. On the following day, the cells were treated with 5 µM of peptides tagged with 5(6)-carboxyfluorescein (peptide-CF) for 3 hours. After 3 hours, the cells are rinsed with PBS and treated with 0.04% of Trypan Blue dye for 5 minutes. Next the wells were washed thrice with PBS and cells were subjected to trypsinization. The cell pellet was then suspended in 200 µL of ice- cold PBS and subjected to flow cytometry analysis [38].
2.8 Cellular uptake of peptides by confocal microscopy
10,000 cells of MDA-MB-231, A549, HeLa and MCF-10A were seeded in 96-well plates and placed in the incubator for 24 hours. The following day, 5 µM of peptide-CF conjugates in serum-free and phenol red-free DMEM and MEBM media was added to the cells for 3 hours in a 37 ºC incubator. Once the incubation period is complete the cells were rinsed twice with PBS and stained with 5 µg/ml Hoechst 33342 stain for 5 minutes. The cells in the 96-well plate were visualized in a confocal laser scanning microscope (Zeiss LSM 980 Airyscan 2) to confirm cellular uptake using 20X objective with 0.8 numerical aperture.
2.9 Colocalization of peptides by confocal microscopy
10,000 cells of MDA-MB-231, A549, HeLa and MCF-10A were seeded in 96-well plates and kept in the incubator for 24 hours. The following day, cells were incubated with 5 µM of peptide-CF conjugates in serum-free and phenol red-free DMEM and MEBM media for 3 hours in a 37 ºC incubator. After 3 hours, two PBS washes were given and incubated with either 125nM of MitoTracker Deep Red dye or 40nM of Lysotracker Deep Red dye for 1 hour. After another PBS wash, the cells were stained with 5 µg/ml Hoechst 33342 stain for 5 minutes. They were visualized in a confocal laser scanning microscope (Zeiss LSM 980 Airyscan 2) to study the organelle (mitochondria or lysosomes) colocalization using 40X objective with 0.95 numerical aperture. Values for Pearson’s correlation coefficient for colocalization was determined using the colocalization module in the Zeiss Zen Blue software.
2.10 Cytotoxicity assay
Cytotoxicity in MDA-MB-231 and MCF-10A cells was determined using the tetramethyl rhodamine methyl ester (TMRM) dye. 10,000 cells were seeded in a 96-well plate and placed in an incubator overnight. The next day, cells were washed once with PBS and stained using 5 µg/ml Hoechst 33342 for 5 minutes. Next, the cells were treated with media containing 100nM TMRM dye and incubated at 37 ºC for 10 minutes. Cells were rewashed with PBS once more. Cells were then treated with peptides conjugated with the drug methotrexate (peptide-MTX) at different concentrations (5, 10, 15 and 20 µM), prepared in media containing 2% FBS and 20nM of TMRM. This was subjected to incubation for 48 hours in an incubator. Fluorescent images using confocal microscope (Nikon A1R HD) were taken in red channel for mitochondrial staining and in blue channel for nuclear staining using 20X objective with 0.75 numerical aperture.
2.11 Apoptosis assay
10,000 MDA-MB-231 cells possessing stable expression of FRET-based caspase sensor (DEVD) were used to determine the apoptosis inducing potential of the peptide-drug conjugates. After seeding cells, peptide-MTX treatment at various concentrations (5, 10 and 15 µM) was given. After incubation for 48 hours, imaging of CFP/YFP FRET ratio was performed using fluorescence microscope (Nikon). The images were analysed to generate quantitative data using the color code of CFP/YFP FRET ratio ranging from scale 0 to 2.
2.12 Serum biocompatibility assay
70,000 cells of MDA-MB-231, A549, HeLa and MCF-10A were cultured in 24-well plates and placed in an incubator at 37 ºC overnight. The following day, 5 µM of peptide-CF conjugates were mixed with equal volume of Fetal Bovine Serum (FBS) and incubated at 37 ºC for 1 hour. After one PBS wash, cells were subjected to treatment with serum-incubated and serum-free peptide-CF for 2 hours. After 2 hours, the cells are washed once and treated with 0.04% of Trypan Blue dye for 5 minutes. Multiple PBS washes were given and cells were subjected to trypsinization. The cell pellet was then suspended in 200 µL of ice- cold PBS and subjected to flow cytometry analysis.
2.13 Hemolysis Assay
2ml of blood was drawn from a healthy individual in a tube containing sodium heparin. The whole blood was centrifuged at 2000 rpm for 5 minutes to separate the plasma from the RBCs. 2ml of Phosphate buffer saline (PBS, pH 7.4) was added to the layer of RBCs and this was subjected to centrifugation at 2000 rpm for 5 minutes. This step of washing with PBS was repeated thrice until a clear supernatant was observed. Next, a 20% haematocrit was prepared from the RBCs by mixing 2 ml of PBS with 1 ml of RBCs. In a microcentrifuge tube, 50 µl of 20% haematocrit was mixed with an equal volume of 40µM peptide-MTX conjugate and subjected to incubation for 2 hours at 37 ºC in a shaking incubator. Complete hemolysis of RBCs and no hemolysis was obtained using 0.5% Triton X-100 and PBS respectively. The tubes were then centrifuged at 800g for 5 minutes and 75µl of the supernatant was added in triplicate to the wells in a 96-well plate. Absorbance of the samples was read at 540 nm wavelength, and hemolysis (percent) was calculated as following [31]-
$$Percent hemolysis\left(\%\right)=\frac{{\text{A}}_{540\text{n}\text{m} }\text{S}\text{a}\text{m}\text{p}\text{l}\text{e} -{ \text{A}}_{540\text{n}\text{m} }\text{N}\text{e}\text{g}\text{a}\text{t}\text{i}\text{v}\text{e} \text{c}\text{o}\text{n}\text{t}\text{r}\text{o}\text{l}}{{\text{A}}_{540\text{n}\text{m} }0.5\text{%} \text{T}\text{r}\text{i}\text{t}\text{o}\text{n} \text{X}100 - {\text{A}}_{540\text{n}\text{m} }\text{N}\text{e}\text{g}\text{a}\text{t}\text{i}\text{v}\text{e} \text{c}\text{o}\text{n}\text{t}\text{r}\text{o}\text{l}}\times 100$$
2.14 Cancer stem cell isolation by Side-population assay
Side-population cells can be isolated using flow cytometry by exploiting their ability to efflux out the DNA-binding dye Hoechst 33342 by ABC transporters. The side-population cells are enriched in cancer stem cells that induce tumors, cause malignancies and resistance to therapy. This technique is very useful to isolate and culture cancer stem cells and study the effect of therapeutic molecules in-vitro [39]. A549 cells were seeded in 100-mm tissue culture plates to reach a confluency of 50–70%. Cells were detached by trypsinization and washed with PBS. 1x106 cells were subjected to Hoechst staining (5 µg/ml) for a duration of 90 minutes in a water bath and shaking the cells intermittently every 15 minutes. One set of cells were also treated with 100µM of the drug Verapamil along with Hoechst staining, and incubated in a similar way. After 1.5 hours, the cells were centrifuged and sorted by flow cytometry. The Hoechst dye is excited with a UV laser at 355 nm and its fluorescence emission is measured with both 505 long-pass 670/40 filter (Hoechst Red) and 450/50 filter (Hoechst Blue). The side-population cells and the main population cells were collected through cell sorting and seeded in 96-well plates.
2.15 Comparison of cellular uptake of peptides in side-population and main-population cells
5000 side-population and main-population cells of A549 were seeded in a 96-well black-coloured clear bottom tissue culture plate. The following day, cells were treated with 5µM peptide-CF conjugates for three hours. Wells were washed with PBS twice and stained with 5µg/ml Hoechst 33342 nucleic acid stain for 10 minutes at 37 ºC. Imaging was performed using confocal laser scanning microscopy (Zeiss LSM 980 Airy Scan 2). The uptake of peptides in side-population and main-population cells was compared by measuring the intensity in the green channel.
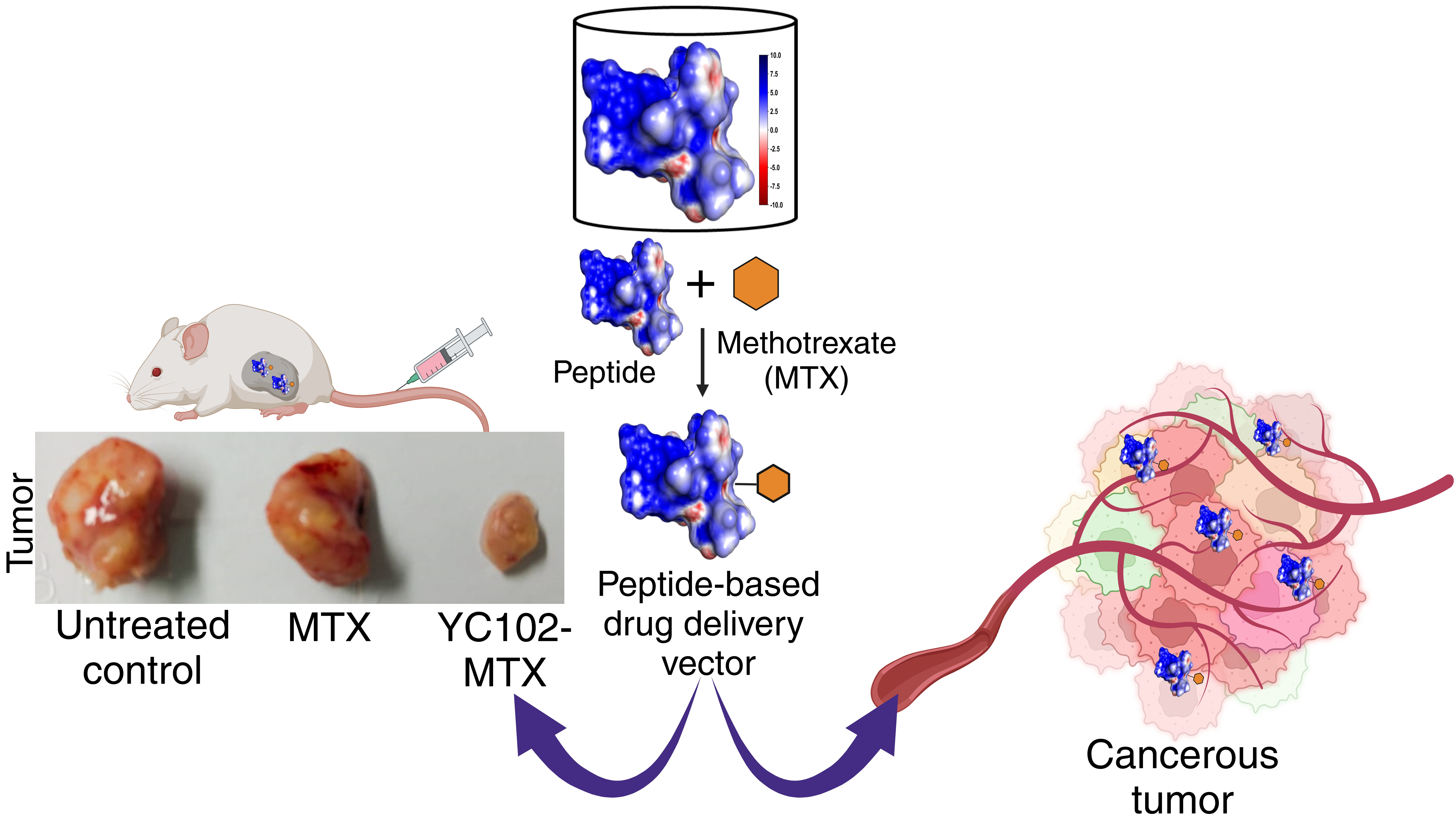
2.16 Tumor regression studies
For in-vivo experiments, NSG (NOD SCID gamma mouse) female mice were maintained at the Animal Research Facility of RGCB in a pathogen free condition. Cells of MDA-MB-231 showing stable expression of Luc2-TdTomato (matrigel 50:50) were injected to the right fourth mammary fat pad in a subcutaneous manner. Tumor formation was confirmed by palpation and mice were segregated into three groups based on the treatment-control, MTX treated and YC102-MTX treated (N = 3). The peptide drug conjugate YC102-MTX, MTX or normal saline were administered as 100µl injection once every 14 days. YC102-MTX and MTX were administrated at a concentration of 2mg/kg body weight of the mice. After the end of 4 weeks, the mice were placed under anesthesia using 2% isoflurane and D-luciferin potassium salts was injected by the intra-peritoneal route. Bioluminescence imaging was performed using IVIS Lumina system (Caliper Life Sciences). Every fifth day the tumor volume was measured using digital caliper. The animals were also euthanized and organs such as lung, spleen, heart, kidney and tumor tissues were collected for histopathology studies.
2.17 Biodistribution assay
To check for the biodistribution in mice, peptide YC102 was tagged with the fluorophore cyanine 7 (Cy7) at the N-terminus. Female NSG mice were segregated in 2 groups, healthy mice and tumor-bearing mice (N = 3). YC102-Cy7 conjugate was administered as 100µL injection through the tail vein at a concentration of 2 mg/kg body weight. Mice were placed under isoflurane anesthesia and imaged at the time intervals of 3, 24, 48, 72, 96 and 120 hours using IVIS Spectrum in vivo imaging system (Perkin Elmer). After 120 hours, the mice were euthanized and the tumor was collected and washed to check for surface fluorescence. Blood from facial veins was also collected from healthy and tumor-bearing mice at regular time intervals of 20 minutes upto 4 hours. Presence of YC102-Cy7 was checked and compared from the blood plasma.
2.18 Histopathology studies
Organs such as liver, lung, spleen and tumor xenograft were collected, sectioned and subjected to H&E staining after performing dewaxing, dehydration and washing. The sections were imaged using microscopy.
2.19 Statistical analysis
All experiments were performed with a minimum sample size of three with data represented as mean ± SEM. Statistical analysis was done using OriginPro software (OriginLab Corporation, USA). Comparison of data between groups was done using using one-way analysis of variance (ANOVA) with significance value set at p-value < 0.05 (*).
{kind=link}