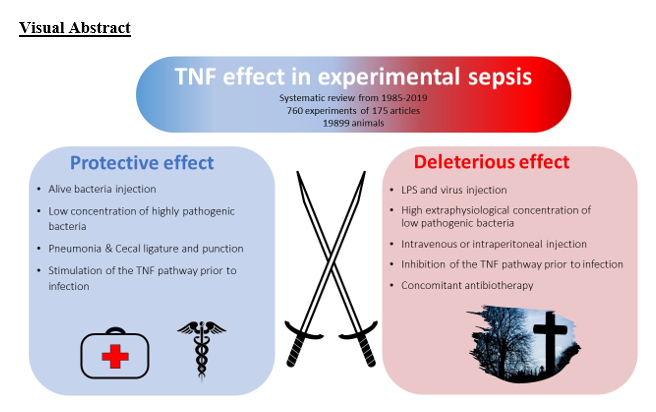
The main finding of this meta-analysis regarding the role of TNF in infection and sepsis in animal models is the strong dependence of this role on various experimental parameters. These parameters include the animal species, their genetic background, the method used for experimental modulation of the TNF pathway, the sequence of injection of modulation agent of the TNF pathway, the type of pathogen, and the infection model used. The variety of these parameters, which may interact in divergent ways, may explain why the majority of experiments (41%) did not provide a clear conclusion regarding the role of TNF.
However, when considering the conclusive experiments, an apparent pattern emerges suggesting that TNF typically exerts a protective role unless the immune system is experimentally overwhelmed by a sudden and intense infectious stimulus (e.g. LPS as a superantigen), in which case TNF appears to be detrimental. Consequently, TNF tends to play a deleterious role in models that induce rapid and high mortality rates, such as infectious models that use very high pathogen loads or direct injections of LPS. These models are rarely representative of the situations encountered in clinical practice, which should serve as the basis for understanding the pathophysiological role of key players. In contrast, TNF appears to play a protective role in models that more closely mimic sepsis scenarios encountered in clinical practice, such as pneumonia or CLP. However, these findings must be interpreted with caution due to their high level of bias and their limited relevance to the standards of good practice expected in clinical trials, such as randomization and blinding.
The results of this systematic review are illustrated by the 1987 study by Tracey et al.[38], which was the first to suggest a deleterious role for TNF using live bacteria (E. coli). In this study, a laboratory strain of E. coli was administered at a dose greater than 1.2x1012 bacteria inoculated intravenously over 30 minutes, in conjunction with early administration of antibiotics. However, this initial inoculum does not appear to be physiological. In fact, feces, the natural element containing the highest concentration of E. coli, typically has concentrations around 108 Enterobacterales per gram[39]. Consequently, the bacterial load used in this study would be equivalent to 10 kg of feces. The authors did not provide any justification for the choice of such a dose, and it is difficult to imagine such a scenario occurring in nature, especially via intravenous administration. In addition, the rapid use of antibiotics makes these models more akin to acute inflammation models, similar to LPS injection, with extremely high TNF levels, exceeding those found in more physiological models such as CLP[40]. For instance, in the 1985 article by Beutler et al.[3], which was the first to suggest a deleterious role of TNF in infections, LPS was progressively injected until lethality was induced. However, the lethal dose was not compared with the circulating levels of LPS found in clinical situations of infection. Other studies have also investigated this subject and found relatively low circulating levels. In their 2003 study, Echtenacher et al.[19] found that 48 hours after a CLP, the concentration of LPS in serum was 0.358 ng/ml in mice. However, after the injection of just 1µg of LPS, the concentration increased to 1,004 ng/ml, which is 3,000 times higher. Remarkably, this dose of LPS did not induce any mortality, with the lethal dose being at least 100 µg/ml, i.e., 300,000 times higher than the concentrations observed in physiological CLP situations. This is in line with studies that evaluate circulating LPS levels in humans. In the first study to measure circulating endotoxins in human sepsis[41], only 17% of patients had endotoxemia, and at relatively low concentrations (5 to 0.5 ng/ml). This result has been confirmed by more recent studies[42, 43].
Additionally, 25% of LPS experiments involve the injection of galactosamine along with LPS. Galactosamine is used to sensitize mice to LPS and reduce the amount needed to cause mortality. Unlike other species such as rabbits, mice are highly resistant to LPS and require large doses to be lethal[44, 45]. However, galactosamine can induce fulminant hepatitis[46] by sensitizing hepatocytes to LPS, resulting in significant apoptotic cell death of nearly all hepatocytes[47]. This mechanism differs from the mortality induced by LPS. Mice resistant to the associated LPS-galactosamine may present the same mortality rate as wild-type mice to LPS injection[48, 49]. As LPS accounts for 40% of the experiments and is the main infectious stimulus assessing the role of TNF, this could explain why this cytokine is often considered to be deleterious in sepsis[50, 51]. Nowadays these limitations of LPS are known, and LPS is no longer recommended for studying sepsis[52–54]. However, sepsis definitions are still partially based on these results. Sepsis is defined as a dysregulated host response to infection[18], whereas experimental data that focused on particularly virulent bacteria found, on the contrary, a protective role of TNF.
In our review, experiments with virulent bacteria, which used much lower initial inocula, appear to more closely reflect the situations observed in clinical contexts. For instance, L. monocytogenes, a pathogen that is well-known for inducing meningitis and maternal-fetal infections, was the primary organism used and was strongly associated with a protective role of TNF. The initial intravenous dose varied between 102 and 105 CFU (104-107 CFU/Kg). However, the threshold for food contamination with L. monocytogenes is set at 100 CFU/g of food[55]. The doses used in these articles corresponded to quantities of contaminated food ranging from 1g to 1kg, which are more realistic scenarios than the bacterial load used with E.coli as previously mentioned[38]. The use of encapsulated bacteria was associated with even lower doses of pathogens. Cross et al.[28] found that less than a dozen encapsulated E. coli bacteria were sufficient to kill all the TNF KO mice, while the control mice died with a bacterial load 105 times higher (5x105 CFU/kg) CFU. In contrast, the same E. coli lacking the capsule required an inoculum of about 107 CFU (5x108 CFU/kg) to induce mortality in all the mice, with no difference observed between the TNF KO and control mice. The capsule appears to cause mortality variations that are equally significant as the absence of TNF. Capsules are commonly present in E. coli bacteria and act as virulence factors that are prevalent in clinical settings, particularly in neonatal meningitis and urinary tract infections[56, 57]. Due to their capsule, these bacteria are resistant to phagocytosis and the complement pathway, particularly in children[58, 59]. These findings are consistent with experiments demonstrating increased invasiveness of encapsulated E. coli compared to non-encapsulated E. coli [57, 60]. Additionally, E. coli encountered in clinical settings can produce toxins such as Shiga toxin, contributing to their high virulence[61, 62]. Furthermore, strains of E. coli responsible for pyelonephritis differ from those causing cystitis[63]. Overall, these results underscore that laboratory bacterial strains are unable to induce the same level of sepsis as the virulent bacteria encountered in clinical settings. A prime example of this is S. pneumoniae, which is the microorganism most commonly found in young and elderly patients. Despite the use of antibiotic therapy and the development of pneumococcal vaccination, pneumococcal infection remains a leading cause of bacterial pneumonia and community-acquired meningitis, and almost 800,000 children under the age of 5 die each year worldwide from this infection. In our review, no study found a deleterious role of TNF in relation to this pathogen. For example, in two studies, a dose of 100 pneumococci injected intraperitoneally was sufficient to kill more than 80% of Tnfr1- KO mice, whereas 107 CFU were required to induce similar mortality in control mice [64, 65]. These results support the notion that TNF plays a protective role in pneumococcal infections, which is consistent with other experimental studies involving animals lacking genes encoding molecules such as IL-1B[66], TLR-4[67] and TLR-2[68], TLR-9[69] and MYD88[70, 71].
When interpreting experiments that report lethal injections of TNF, the dose must also be considered. In the 1987 study by Tracey et al., the administered dose of TNF required to induce mortality in 50% of the rats was 0.6mg/kg, resulting in a peak plasma TNF concentration of 600ng/ml. In the study by Rothe et al., the dose required to induce mortality in 50% of the mice was 1µg per mouse, equivalent to 0.05mg/kg, resulting in an estimated peak plasma TNF concentration of 66 ng/ml[31].
Feuerstein et al[72] found that injection of recombinant TNF at concentrations 10–100 times higher (107 UI/ml) than those found in rats succumbing to LPS injection did not result in lethality. These levels of circulating TNF are significantly higher than those found in septic patients, which typically do not exceed 0.1ng/ml4,11,57,58. Some experimental models based on E. faecalis[75, 76], with mortality rates exceeding 50%, have failed to detect measurable levels of TNF. Furthermore, in the 1989 study by Sheppard et al., the injection of 50 µg/ml TNF in rats, resulting in a peak concentration of approximately 50 ng/ml, was not deleterious but rather protective, shielding the animals from a lethal injection of LPS administered 24 hours later[77]. In the same year, Hershman et al.[78] conducted a study wherein the administration of 0.1µg of recombinant TNF, resulting in an estimated peak concentration of 6 ng/ml, exhibited a protective effect by increasing the survival rate of mice with K. pneumoniae skin infection. These experimental findings suggest that very high levels of TNF, well above those typically observed in septic patients, may not only be compatible with life but may also confer a protective effect during infection. This phenomenon may be related to increased secretion of soluble TNF receptors, which may attenuate the increase of TNF, except during the very early stages of infection[79]. This may help elucidate why certain studies in the mid-1990s showed that circulating TNF levels were not correlated with mortality and could even be protective in human sepsis[80, 81].
Our meta-analysis has three major limitations. Firstly, almost 6% of the eligible publications could not be analyzed because the articles could not be found. However, half of these articles were not written in English and may have little impact on the current opinion. Secondly, although we aimed to be as comprehensive as possible, our literature search strategy was unable to include all articles where both the TNF signaling pathway was altered and mortality was assessed. However, this investigation, which includes 175 studies, represents the largest meta-analysis on this topic to date. Thirdly, this meta-analysis presents a high risk of bias, as most of the studies included were neither randomized nor blinded. However, there appears to be a general consistency across the studies. None of them indicated that injecting low doses of LPS or virulent microorganisms, such as encapsulated bacteria, was deleterious. On the other hand, ten of the studies included highlighted the dual role of TNF. It was found to be protective in the case of infection, but deleterious when LPS was administered (Table 5). The contrasting role of TNF in response to LPS and E. coli injection compared to more virulent pathogens such as S. pneumonia and Listeria spp was also highlighted in a systematic review conducted in 2005[82]. However, this review exclusively focused on studies involving TNF neutralization and exhibited several methodological limitations. First, the definition of sepsis was not standardized and included infections caused by microorganisms such as Toxoplasma gondii or mycobacteria, commonly associated with chronic infections. In addition, the statistical analysis was restricted to assessing the reduction in absolute risk of mortality without using a random-effects model, as recommended in meta-analysis protocols[17]. Furthermore, a study from 1996 highlighted the differential effects of various anti-inflammatory drugs between Gram-negative and Gram-positive bacteria. In this study, TNF blockade using soluble TNF receptor 2 was associated with a protective effect in Gram-negative infections, whereas it proved deleterious in Gram-positive infections[83].
{kind=link}