2.1 Synthesis, isolation and characterization:
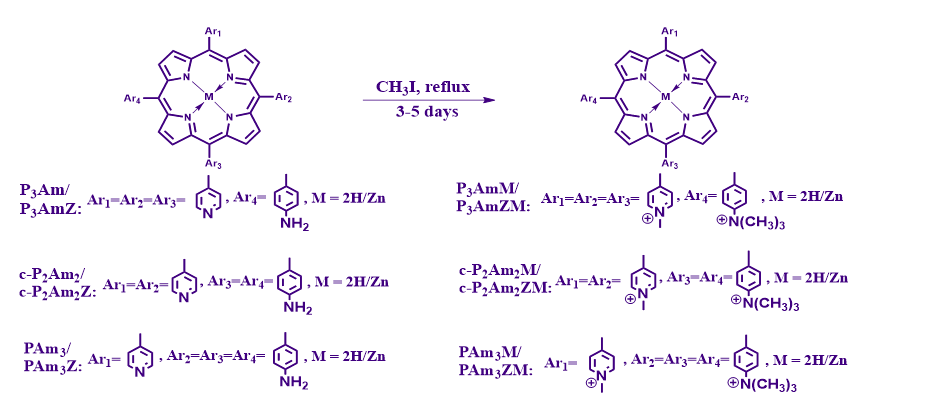
2.1.1 Synthesis: The cationic water-soluble porphyrins were synthesised through a multi-step methodology as outlined in Scheme S1. A one-pot three-component reaction involving 4-acetamidobenzaldehyde (1.5 eq), pyridine-4-carboxaldehyde (3.2 eq) and pyrrole (4 eq) led to the formation of the products 5,10,15-tri-(4-acetamidophenyl)-20-(4-pyridyl) porphyrin (PA3), 5,10-di-(4-acetamidophenyl)-15,20-di-(4-pyridyl) porphyrin (c-P2A2), 5-(4-acetamidophenyl)-10,15,20-tri-(4-pyridyl) porphyrin (P3A). TLC of the crude reaction mixture indicated the formation of six compounds, of which only three (PA3, c-P2A2 and PA3) were isolated and characterised. Isolation was achieved through gravity percolation chromatography, silica gel 60–200 mesh and (3%-10%) MeOH in DCM being the stationary and mobile phases, respectively. P3A was the first to elute with 5% MeOH in DCM followed by c-P2A2. PA3 eluted out in 7% MeOH in DCM. Wherever required, a second column was run for further purification. P3A, c-P2A2 and PA3 were obtained in 10%, 9% and 6% yield. The isolation of the rest of the fractions was not attempted. The compound P3A has been reported earlier, the synthesis being achieved through a protocol either similar to the one included in this paper41 or a different one.42,43 Compound c-P2A2 has been reported earlier with a different synthetic strategy.44 To our knowledge, PA3 has not been reported so far. The acetamidophenyl porphyrins were then hydrolysed (Scheme S1, SI) to form the corresponding meso-aminophenyl substituted porphyrin derivatives 5,10,15-tri-(4-aminophenyl)-20-(4-pyridyl) porphyrin (PAm3), 5,10-di-(4-aminophenyl)-15,20-di-(4-pyridyl) porphyrin (c-P2Am2), 5-(4-aminophenyl)-10,15,20-tri-(4-pyridyl) porphyrin (P3Am). As shown in Scheme S2, an ethanolic solution of P3A, e.g., is reacted with HCl (5 M) under reflux conditions, the amide bond of the lone acetamidophenyl group of the compound gets cleaved, forming an aminophenyl function as in compound P3Am. PAm3, c-P2Am2 and P3Am were obtained in 82%, 96% and 88% yield, respectively. The synthesis of P3Am with the same reagent mixture but under different conditions of reaction and workup has been reported earlier.41,45 In contrast compounds c-P2Am2 and PAm3 have been synthesised using an entirely different methodology involving a Lossen rearrangement. Derivatives of the type CMxPyyP were warmed to 100°C in polyphosphoric acid, then gradually to 160°C over 3 h followed by neutralisation with NaOH to obtain c-P2Am2 and PAm3 (or cis-A2Py2P and APy3P) as reported in the publications.42,43 Zn-metalation of the freebase porphyrins P3Am, c-P2Am2 and PAm3 was carried out by reacting the individual compounds with zinc acetate in CHCl3/MeOH-9:1 v/v solution, as shown in Scheme S3. The Zn-metal complexes P3AmZ, c-P2Am2Z and PAm3Z were obtained in 87, 86 and 78% yields, respectively.
The freebase porphyrins P3Am, c-P2Am2, PAm3 and the zinc complexes P3AmZ, c-P2Am2Z and PAm3Z were then alkylated through a room temperature reaction with methyl iodide to produce the hydrophilic poly iodide salts P3AmM, c-P2Am2M, PAm3M and their Zn metal complexes P3AmZM), c-P2Am2ZM and PAm3ZM (Scheme 1). In each case, alkylation of the meso-(4-pyridyl) groups to meso-(4-methylpyridinium) and meso-(4-aminophenyl) to meso-(4-trimethylammoniumphenyl) moieties was achieved. This synthetic achievement results in the first-ever report of direct quaternarisation of meso-(4-aminophenyl) moieties in porphyrins bearing combination of meso-(4-aminophenyl) and meso-(4-pyridyl) groups. The synthesised polyiodide salts were readily hydrophilic, thus rendering them useful for biochemical explorations. Alkylation involving 5-(4-aminophenyl)-10,15,20-tri-(4-pyridyl) porphyrin attempted with a different synthetic methodology has been reported earlier.46 Nevertheless, the authors could not achieve quaternarisation of the meso-(4-aminophenyl) group.
2.1.2 Characterisation: The synthesised compounds were characterised using 1HNMR, HRMS (ESI)/MALDI-TOF, UV-Vis, and emission spectroscopic techniques. The proton splitting pattern of the synthesised compounds has been included in Table S1 for greater clarity. The splitting patterns are consistent with the A3B/AB3 and cis-A2B2 systems reported in the literature.8,9,47–50 For compounds P3A, c-P2A2, and PA3, the spectral plots (Figure S5-S7) indicate that the β-pyrrole protons are split into doublet-singlet-doublet at 8.94 (d, J = 4.7 Hz)−8.85 (s)−8.82 (d, J = 4.7 Hz) ppm, 8.92 (d, J = 4.7 Hz)−8.89 (s)−8.83 (s)−8.8 (d, J = 4.7 Hz) ppm, and 8.92 (d, J = 4.2 Hz)-8.89(s)−8.84 (d, J = 4.2 Hz) ppm, respectively. The pyridyl protons resonate at 9.05 (d, J = 4.1 Hz), 8.16 (d, J = 4.1 Hz) for P3A, 9.05 (d, J = 5 Hz), 8.16 (d, J = 5 Hz) for c-P2A2 and 9.04 (d, J = 4.9 Hz), 8.27 (d, J = 4.9 Hz) ppm for PA3.
The proton splitting for the phenyl protons appeared at 7.94 (d, J = 7.9 Hz) and 7.56 (d, J = 7.9 Hz) for P3A, 7.92 (d, J = 7.9 Hz) and 7.53 (d, J = 7.9 Hz) for c-P2A2, 8.15 (d, J = 8.1 Hz) and 8.06 (d, J = 8.1 Hz) ppm in the case of PA3. The 1HNMR spectral data of P3A conforms to the published values.41,45,51 For P3Am, c-P2Am2 and PAm3, the 1HNMR spectral plots (Figure S8-S10) and tabulated spectral data (Table S1) agree with the published values42,43 and confirm the identity of the compounds. The HRMS (ESI) data of the acetamidophenyl and aminophenyl porphyrins further indicate their purity; the mass error in each case was less than 5 ppm (Figure S20-S25). The striking feature in 1HNMR spectral data (Table S1 and Figure S11-S13) of the Zn-complexes is the disappearance of the highly shielded inner pyrrolic proton peak, a characteristic of the successful metalation of the porphyrins. Apart from that, the β-pyrrole splitting patterns (Fig. 2) conform to that of the A3B/AB3 and c-A2B2 systems. For P3AmZ and PAm3Z, the β-pyrrole protons are split into doublet-singlet-doublet centred at 8.98 (d, J = 4.4 Hz)-8.81 (s)-8.79 (d, J = 4.4 Hz) and 8.9 (d, J = 4.6 Hz)-8.88 (s)-8.71 (d, J = 4.6 Hz) ppm, respectively. Proton resonances at 8.94 (d, J = 4.4 Hz)-8.91 (s)-8.77 (s)-8.74 (d, J = 4.4 Hz) ppm were observed for β pyrrolic protons of c-P2Am2Z. The pyridyl and phenyl protons are split into four doublets centred at 9.01 (d, J = 4.6 Hz), 8.21 (d, J = 4.6 Hz) and 7.83 (d, J = 8.1 Hz), 6.99 (d, J = 8.1 Hz) ppm, respectively
for P3Am, 8.98 (d, J = 4.9 Hz), 8.19 (d, J = 4.9 Hz) and 7.83 (d, J = 8.1 Hz), 6.99 (d, J = 8.1 Hz) ppm, respectively for c-P2Am2Z and 8.97 (d, J = 4.9 Hz), 8.19 (d, J = 4.9 Hz) and 7.82 (d, J = 7.8 Hz), 6.98 (d, J = 7.8 Hz) ppm, respectively for PAm3Z.
The amino-protons resonated as a singlet at 5.53 (s, br), 5.55 (s), and 5.48 (s) ppm for P3AmZ, c-P2Am2Z, and PAm3Z, respectively. The formation of the compounds was ably supported by MALDI-TOF data (Figure S26-28). The observed and calculated mass were in good agreement with each other.
The alkylation of the free-base compounds and their Zn-complexes was supported by the recorded MALDI-TOF (Figure S29 – S34) spectra, the mass values obtained agree well with the calculated values, and the mass error is within 5 ppm limit. 1HNMR (Figure S14 – S19) gives further evidence in support of the alkylation, intense singlets corresponding to −CH3 protons in methylpyridinium and ammoniumphenyl moieties were consistently observed. However, the aromatic proton splitting presented a rather complex picture. This observation may be because of the effect of the cationic charges on electrons around the porphyrin ring. Protonation of aminophenyl porphyrins forming the ammoniumphenyl counterparts has been reported to have affected 1HNMR spectra.43
The synthesised porphyrins' UV-Vis spectra (Figure S1 A, S2 A, S3 A) consistently depict the Soret and Q band absorptions. The absorption peaks, molar extinction coefficient, emission peaks and Stokes shift have been included in Table S2. Minor redshifts in the absorption peaks were observed for P3A, c-P2A2 and PA3 (Figure S1 A). Emission spectra (Figure S1 B) recorded through excitation of the same solutions of the compounds used for UV-vis measurement at the absorption maxima of the respective Soret band exhibit peaks at 644, 660, 717 nm for P3A, 647, 664, 720 nm for c-P2A2 and 646, 673, 723 nm for PA3.
For the aminophenyl porphyrins P3Am, c-P2Am2 and PAm3 redshifts in Soret band absorptions were apparent (Figure S2 A) as the number of meso-aminophenyl groups increased in the porphyrin skeleton in going from P3Am−c-P2Am2−PAm3, the magnitude of redshift being 6 nm in going from P3Am to c-P2Am2 and 5 nm between c-P2Am2 and PAm3. Redshifts to the tune of 20 nm can also be seen in the emission profile (Figure S2 B) of compounds P3Am, c-P2Am2 and PAm3 with a progressive increase in the number of meso-aminophenyl groups. The emission peaks corresponding to the Q(0,0) and Q(0,1) transitions are 653 (\({{\lambda }}_{\text{e}\text{m}}^{\text{m}\text{a}\text{x}}\)), 718 nm for P3Am, 673 (\({{\lambda }}_{\text{e}\text{m}}^{\text{m}\text{a}\text{x}}\)), 723 nm for c-P2Am2 and 694 (\({{\lambda }}_{\text{e}\text{m}}^{\text{m}\text{a}\text{x}}\)), 757 nm for PAm3, although it should be stated that the Q(0,1) emission band is not well defined in c-P2Am2 and PAm3 and it appears more like a shoulder peak in the spectral plots (Figure S2 B). Stokes shift of 237, 251 and 263 nm were observed for P3Am, c-P2Am2 and PAm3, the values are much lower than the acetamidophenyl porphyrins with Stokes shift of 299, 300 and 301 nm for P3A, c-P2A2 and PA3.
Evidence of Zn-metalation can be seen in the UV-Vis spectral plot (Figure S3 A) of the Zn-complexes. The incorporation of Zn is accompanied by the reduction in the number of Q-band absorptions from four in the freebase porphyrin to two in the metal complex. The reduction in the number of Q-band peaks in the Zn porphyrins is a direct consequence of the increased symmetry of the compounds relative to that of the free base porphyrins. The inner pyrrolic protons in the porphyrin core reduce the symmetry from D4h to D2h.52 Redshift in Soret band maxima can be seen with an increase in the number of appended aminophenyl groups. The λmax of the Soret band absorption for P3AmZ, c-P2Am2Z and PAm3Z are 427, 432 and 435 nm, respectively.
The Q-band peaks appear at 560 and 603 nm for P3AmZ, 563 and 607 nm for c-P2Am2Z and finally, 566 and 611 nm for PAm3Z. A blueshift of emission maxima also accompanies complex formation (Figure S3 B); the compounds also show a blue shift in Q(0,0) emission peaks with a progressive reduction in the number of meso-aminophenyl groups from three in PAm3Z to two in c-P2Am2Z and to just one in P3AmZ. Blueshifts were also observed in the Q (0,1) emission band with a reduction in the number of meso-aminophenyl groups. However, the trend is different, with the observed blue shift being highest for c-P2Am2Z.
The comparative UV-Vis plot of the compounds (Fig. 3) clearly shows a blue shit in Soret band absorption of the meso-acetamidophenyl porphyrins upon hydrolysis to meso-aminophenyl porphyrins. Zn-metalation of the latter was accompanied by a significant red shift in λmax of the Soret band. Table S2 includes the detailed UV-Vis and emission data of P3A, c-P2A2, PA3, P3Am, c-P2Am2, PAm3, P3AmZ, c-P2Am2Z and PAm3Z with calculated Stokes shift and molar extinction coefficient for better clarity. The Stokes shift values show a progressive reduction in going from the freebase acetamidophenyl porphyrins to the amino porphyrins and finally to the Zn complexes.
Such a decrease of Stokes shift in Zn-complexes relative to the freebase system was reported earlier53,54 and can be attributed to the Zn-compounds taking on a more co-planar configuration in the first excited state than in the ground state.53 Between the acetamidophenyl porphyrins and their hydrolysis products, the meso-(4-acetamidophenyl) groups probably induce more significant structural distortions on photoexcitation and, thus, the larger values of Stokes shift.
The UV-Vis spectra of the CPs were recorded with a 10 µM solution of the compounds in water. The spectral plot and data are presented in Fig. 4 and Table S3. The absorption spectra of the free-base CPs conform to the typical porphyrinic spectra exhibiting an intense Soret band and four Q-band absorptions. The Soret band peaks are redshifted in the Zn complexes compared to their freebase counterpart. The number of Q-bands in the zinc complexes is also reduced to two. The observed spectral behaviour is in line with Gouterman’s model.55
A progressive red shift in the Soret band is also apparent in going from PAm3ZM-c-P2Am2ZM-P3AmZM-TMPyPZ (Fig. 4A). The emission spectra (Fig. 5) of the ammoniumphenyl porphyrins show variable results. The spectral data were recorded by excitation of a 10 µM solution of the compounds in water at the λmax of their Soret bands. While emission peaks pertaining to the Q(0,0) and Q(0,1) emission bands can be observed for P3AmZM, c-P2Am2ZM with emission peaks at 630 (\({{\lambda }}_{\text{e}\text{m}}^{\text{m}\text{a}\text{x}}\)), 653 nm and 634, 654 (\({{\lambda }}_{\text{e}\text{m}}^{\text{m}\text{a}\text{x}}\)) nm, respectively, the two peaks appear to have merged for PAm3ZM with a maximum at 657 nm. The freebase CP c-P2Am2M also exhibits two emission bands at 658 and 710 (\({{\lambda }}_{\text{e}\text{m}}^{\text{m}\text{a}\text{x}}\)) nm. For P3AmM a single emission band could be observed with a maxima at 623 nm, whereas for PAm3M emission peaks appeared at 658 (\({{\lambda }}_{\text{e}\text{m}}^{\text{m}\text{a}\text{x}}\)) and 707 nm. The UV-Vis, emission spectral data, and molar extinction coefficient have been included in Table S3. A blue shift in emission peak maxima is apparent on Zn-complexation; however, the emission maxima of P3AmM was blue-shifted by 10 nm compared to its Zn-complex P3AmZM. The Stokes shift calculated56,57 presented interesting outcomes (Table S3). The large values indicate structural changes upon photoexcitation.53,58,59 The Stokes shift values for the freebase porphyrins were more prominent than their Zn-complexes, PAm3M and PAm3ZM being exceptions here. This agrees with earlier reports,53,54 and indicates that the Zn-complexes take on a more planar configuration in the first excited state than in the ground state compared to the freebase compounds.53,59 The large values of Stokes shift also mean reduced homo-FRET and consequently minimal self-quenching.60,61 This is desirable as far as the applications of the CPs as PSs are concerned.
2.4 Molecular docking Outcomes:
The docking simulations conducted on HIV target proteins indicate that compounds c-P2Am2ZM, PAm3M, P3AmZM, P3AmM, PAm3ZM, and c-P2Am2M demonstrate superior binding affinity and interaction profiles. Notably, among all the targets examined, these compounds exhibited stronger binding energy with reverse transcriptase compared to gp120 and protease. Specifically, when targeting reverse transcriptase, all compounds displayed better binding energy than stavudine and 1l4 (Fig. 11). Furthermore, c-P2Am2ZM and c-P2Am2M exhibited superior binding energy with protease compared to other targets. Interestingly, c-P2Am2ZM emerges as one of the top three compounds across all three explored HIV targets. Additionally, PAm3M stands out as a top compound with gp120 and reverse transcriptase proteins.
In the docking program's validation process, Y2E was meticulously redocked into its co-crystallized site on the HIV gp120 protein. A notably low Root Mean Square Deviation (RMSD) value of 1.53 Å was noted between the docked and native poses. This result serves as compelling evidence of the program's ability to accurately predict the binding poses of inhibitors within HIV proteins. This demonstrates the program's capability to reproduce the spatial positioning of Y2E within its designated binding site, affirming the docking algorithm's reliability and precision. The binding energies and molecular interaction profiles of the compounds were compared with those of co-crystallized ligands and drugs targeting various HIV targets. These targets play crucial roles in different stages of the viral life cycle: viral entry (gp120), production of infectious virions (protease), and replication (reverse transcriptase).
Figure 11
Binding energy details of compounds, drugs and co-crystallized ligands.
Binding energy and interaction summary of the compounds with HIV proteins.
Table 1
Molecular interaction summary of top compounds with HIV gp120.
|
Compounds
|
Binding Energy (K.cal/mol)
|
Interacting Amino acids
|
Nature of interactions
|
|
c-P2Am2ZM
|
-8.2
|
ILE371, MET426, GLU370, ASP368, ARG432, VAL430, SER375, ASN425, THR283
|
Amide- π stacked, π- sigma, π-alkyl, π- cation, π- anion, carbon hydrogen bond, van der waals
|
|
PAm3M
|
-8.1
|
ILE371, GLY473, ASP477, ASP474, GLN428, VAL430, ASP368, GLY431, ASN425
|
Amide- π stacked, π- sigma, π-alkyl, π-anion, π- anion, carbon-hydrogen bond, van der Waals
|
|
P3AmZM
|
-7.9
|
ILE371, ASP474, ASP477, ALA281, GLY473, GLN428, GLY431, VAL430, ASN425, ASP368
|
Amide- π stacked, π- sigma, π-alkyl, π-anion, π- anion, carbon-hydrogen bond, van der Waals
|
Table 2
Molecular interaction summary of top compounds with HIV protease
|
Compounds
|
Binding Energy (K.cal/mol)
|
Interacting Amino acids
|
Nature of interactions
|
|
c-P2Am2ZM
|
-7.7
|
ASP25, LEU23, ARG8, VAL82, PRO81
|
π- cation, π-alkyl, carbon hydrogen bond, van der waals
|
|
c-P2Am2M
|
-7.5
|
ASP25, LEU23, ARG8, VAL82, PRO81
|
π- π-cation, π-alkyl, carbon-hydrogen bond, van der Waals
|
|
P3AmM
|
-6.1
|
ASP30, ASP29, ALA28, ARG8, VAL82, LEU23
|
π- anion, π- cation, π-sigma, π-alkyl, carbon-hydrogen bond, van der Waals
|
Figure 14
3D molecular representation of interactions of compounds A) c-P2Am2ZM, B) c-P2Am2ZM, and C) P3AmM with the active site residues of HIV protease. Interactions were displayed as color-coded dashed lines.
Table 3
Molecular interaction summary of top compounds with HIV reverse transcriptase
|
Compounds
|
Binding Energy (K.cal/mol)
|
Interacting Amino acids
|
Nature of interactions
|
|
PAm3ZM
|
-8.6
|
ARG72, LYS66, LEU74, TYR115, MET184, ALA114, ASP185, PHE160
|
π- π T-shaped, π-alkyl, π- cation, π-sigma, carbon hydrogen bond, van der waals
|
|
PAm3M
|
-8.0
|
ARG72, LYS66, LEU74, GLY152, MET184, ALA114, ASP185, TYR115
|
π- π T-shaped, π-alkyl, π- cation, π-sigma, carbon hydrogen bond, van der waals
|
|
c-P2Am2ZM
|
-7.6
|
ARG72, LYS66, LEU74, ASP185, TYR115, MET184, ALA114
|
π- π T-shaped, π-alkyl, π- cation, π-sigma, carbon hydrogen bond, van der waals
|
The top compounds primarily engaged in hydrophobic and electrostatic interactions, including Amide-π stacked, π-sigma, π-alkyl, π-cation, π-anion, carbon-hydrogen bond, and van der Waals interactions. The interaction profiles, along with the involved residues and types of interactions, are summarized in Tables 1, 2, and 3. Detailed 3D representations of these interactions can be observed in Figs. 12, 14, and 16. Additionally, the binding orientations within the active site pockets can be visualized in Figs. 13, 15, and 17 for HIV gp120, protease, and reverse transcriptase, respectively.
c-P2Am2ZM displayed a distinct binding orientation compared to other top compounds by positioning its trimethyl amino group deeper within the active site pocket. In contrast, the other two compounds were situated on the outer periphery of the gp120 active site pocket. This distinction in positioning can be observed in Fig. 15. Both c-P2Am2ZM, c-P2Am2M, and P3AmM exhibited a consistent orientation within the protease active site cavity. They projected their trimethyl amino groups deeper into the pocket to establish interactions with crucial active site residues. This observation is illustrated in Fig. 15. However, all the top compounds aligned well within the reverse transcriptase active site pocket, as depicted in Fig. 17.
Across the spectrum of HIV protein targets, including gp120, protease, and RT, the porphyrin analogue t-P2Am2M, characterized by a trans configuration of groups, consistently demonstrated lower binding energies compared to its cis counterparts. The consistent decrease in binding efficacy observed with the trans arrangement emphasizes the critical role of the spatial orientation of molecular groups within the porphyrin structure in effectively targeting HIV protein sites.
For details regarding the binding energy and corresponding molecular interactions of t-P2Am2M0, please refer to the supplementary information (Table S9 & Figure S4).
{kind=link}