Materials
Branched polyethyleneimine (PEI, 25 kDa) was obtained from Sigma-Aldrich (MO, USA). D, L-Lactide (DLLA) was obtained from Alfa Aesar (MA, USA). The anhydrous dimethyl sulfoxide (DMSO) was purchased from J&K Scientific Ltd. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)2000] (DSPE-mPEG) was obtained from A.V.T.(Shanghai) Pharmaceutical Co., Ltd. Curcumin (Cur) was purchased from Alfa Aesar (MA, USA). The 0.8 µm polycarbonate membrane was purchased from Millipore Co. (MA, USA). The mimics of siCCAT1 or cyanine-3 (Cy3) labeled siCCAT1 were obtained from RiboBio Co. (Guangzhou, China). Antibodies of western bolt were purchased from Cell Signaling Technology, Inc and Abcam plc. RNase-free deionized water was purchased from TIANGEN Biotech Co. Ltd (Beijing). LysoTracker Green was obtained from Molecular Probes Inc. (Eugene, OR). The cell counting kit-8 (CCK-8) was obtained from Dojindo Molecular Technologies, Inc., (Japan). Fetal bovine serum (FBS)was obtained from Wisent Inc. 0.25% trypsin–EDTA, RPMI 1640 medium and penicillin/streptomycin were obtained from Thermo Fisher Scientific (MA, USA). The water used was of ultrapure grade and was supplied by a Milli-Q purification system of Millipore Co. (MA, USA).
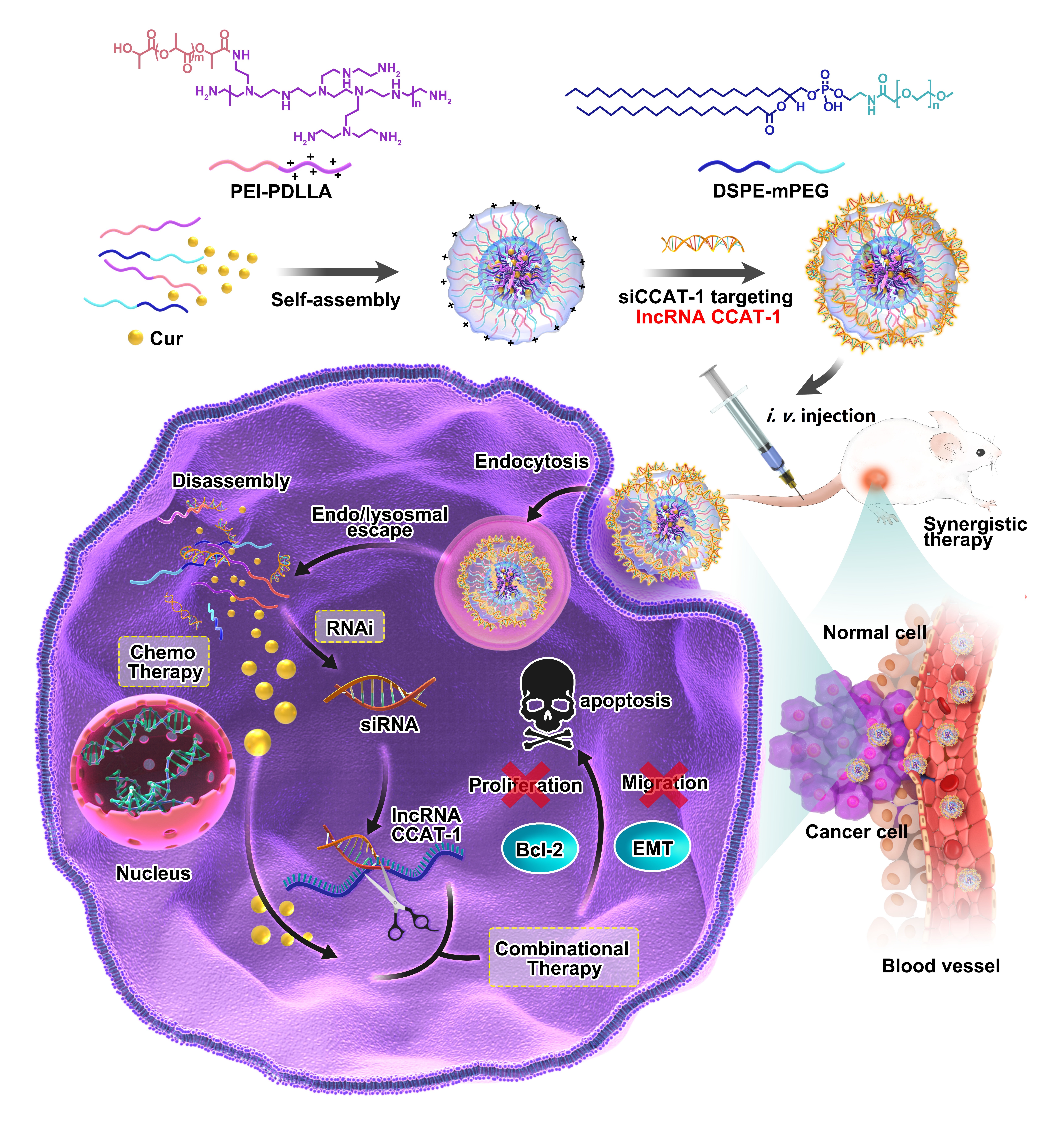
Synthesis of PEI-PDLLA copolymers
Pre-dehydrated 15 g of DLLA and 250 mg of PEI were dissolved in 50 mL of anhydrous DMSO. Then 0.05 mol of trimethylamine was added into above solution, and kept the solution stirring under nitrogen for 12 h at 86℃. The reaction solution was then poured into ice water, and the precipitate was collected and washed by distilled water. Finally, the precipitate was dried in a vacuum oven. The chemical structure was characterized by Fourier transform infrared spectroscopy and proton magnetic resonance spectroscopy.
Preparation of Cur-siCCAT1 co-loaded nanoparticles (CSNP)
Amounts of PEI-PDLLA, DSPE-mPEG and Cur were dissolved into dichloromethane (DCM) to form 5 mg/mL, 5 mg/mL and 10 mg/mL solutions, respectively. The blank nanoparticles (NP) were obtained by mixing PEI-PDLLA and DSPE-mPEG solutions in various ratios and then rotating evaporation by film dispersion method. Briefly, after mixing the two solutions, a thin film was formed in the round-bottomed flask by a rotary evaporator, and then distilled water was added to form NP by ultrasonic vibration for 0.5 h. The mixtures were prepared with PEI-PDLLA: DSPE-mPEG mass ratios of 5: 5, 5: 10, 5: 20, 10:10 and 10: 20.
The Cur-loaded NP (CNP) with different mass ratios of PEI-PDLLA, DSPE-mPEG and Cur was constructed by mixing Cur solution with PEI-PDLLA and DSPE-mPEG solutions together, and then prepared with film dispersion technique as described above. Separation of unloaded Cur by filtering the suspension through a 0.8µm polycarbonate membrane. The siCCAT1-loaded NP (SNP) was formed by adding different mass ratios of siCCAT1 to NP through electrostatic interaction. The mixture of NP and dissolved siCCAT1 were incubated for 30 min at 25℃. The Cur-siCCAT1-co-loaded NP (CSNP) was prepared by adding different mass ratios of siCCAT1 to CNP.
Physicochemical characterization
The average size and zeta potential were detected by dynamic light scattering (DLS) using ZetaSizer Nano series Nano-ZS (Malvern Instruments Ltd, Malvern, UK). After the sample was properly diluted in distilled water, three replicates were analyzed per batch. The morphology of CNP was determined by transmission electron microscopy (TEM) (Ht-7700; HITACHI, Japan). The absorption spectra were analyzed by a UV–vis spectrophotometer (TU-1810; PERSEE, China) and the fluorescence spectra were recorded on a EnSpire® Multimode Plate Reader (PerkinElmer, Fremont, CA).
A UV-Vis spectrophotometer (TU-1810; PERSEE, China) was used to measure the encapsulation efficiency (EE) and load capacity (LC) of Cur at 425 nm. EE and LC were computed by the following formulas:
EE% = (weight of loaded drug) / (weight of originally added drug) ×100%
LC% = (weight of loaded drug) / (total weight of nanoparticles and drug) × 100%
Gel electrophoresis and serum stability assay
CSNP was analyzed by 4% agarose gel electrophoresis. The 4% agarose gel was prepared with tris-acetate-ethylenediaminetetraacetic acid (TAE) buffer containing 0.5 µg/mL Genecolor (Gene-bio, China). For electrophoretic mobility analysis, incubated the sample for 30 minutes at room temperature, and then 10% glycerol was added to the samples. The gel electrophoresis was conducted at 110 V for 10 min, and then the gel was photographed using a Bio-Rad ChemiDoc™ XRSTouch Imaging System (CA, USA).
For the determination of serum degradation, the naked siCCAT1 aqueous solution and CSNP samples were mixed with FBS, at a ratio of 1: 1 to obtain a serum concentration of 50%. The mixtures were then incubated at 37 ℃ for the indicated times. The mixtures were taken out at indicated time interval and then mixed with 2% sodium dodecyl sulfonate (SDS), and which were further loaded onto a 4% agarose gel for gel electrophoresis assay.
In vitro self-tracking of cellular uptake and endosomal/lysosomal escape
The CRC cells HT-29 were cultured in RPMI 1640 medium containing 10% (v/v) FBS and 1% penicillin/streptomycin (P/S) at 37 ℃ in a humidified atmosphere with 5% CO2. HT-29 cells were cultured onto a glass bottom dish, reaching a suitable concentration of 2 × 105 cells per well. The medium was replaced by fresh medium compromising SNP for incubation 0.5 h, 1 h, 2 h, 4 h, 6 h, respectively. After that, HT-29 cells were washed 3 times with PBS, and then stained endosome/lysosome with LysoTracker Green. And cell uptake and endosomal/lysosomal escape of SNP (blue fluorescence) carrying Cy3 labeled siCCAT1 (red fluorescence) and LysoTracker Green labeled endosome/lysosome (green fluorescence) was detected by confocal laser scanning microscope (CLSM, Z-760; Carl Zeiss, Germany).
In vitro cytotoxicity assays
HT-29 cells were seeded in 96-well plates at a density of 5 × 103 cells per well and cultured for 24 h before subsequent studies. Replaced the current medium with 100 µL of RPMI 1640 containing different equivalent density of PBS, naked siCCAT1, NP, free Cur, CNP, SNP and CSNP for 24 h or 48 h (different preparations containing an equivalent 10 µg/mL of Cur or 100 nM of siCCAT1). The cell viability was estimated using the CCK-8 assay according to manufacturer’s instructions (Dojindo, Japan).
To calculate 50% inhibitory concentration (IC50), HT-29 cells in 96-well plates were co-cultured with free Cur in concentrations ranging from 5 to 75 µg/mL. The IC50 was reckoned based on the dose of Cur that caused 50% cell death compared to the PBS control.
Cell apoptosis assay
FITC Annexin V/Propidium Iodide (PI) double staining method was used to determine the ability of inducing apoptosis by different treatment. HT-29 cells co-cultured with a variety of NP, free Cur, CNP, SNP and CSNP were evaluated. After 24 h of incubation, by centrifuging at 500g for 5 min to harvest cells. The Annexin V/PI Apoptosis Detection Kit was used in accordance with the manufacturer ’s protocol and subjected to flow cytometry analysis (FCM).
Western blot assay
Total protein samples in HT-29 cells were extracted with RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China), and then the protein concentration was detected using BCA kit (Beyotime Biotechnology, Shanghai, China). The same amount protein samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). After transferring it to PVDF membrane (Millipore, USA), the protein was incubated in blocking solution (5% skim milk powder) at room temperature for 1 h. The primary antibody β-actin, Bcl-2, Caspase-3, E-Cadherin and N-Cadherin were diluted as 1: 2000 and incubated with the sample at 4℃ overnight, and then reacted with the secondary antibody labeled with horseradish peroxidase for 1 h. The Bio-Rad ChemiDoc™ Touch Imaging System was used to determine protein levels in cells, with β-actin as an internal reference.
In vitro wound healing assay
HT-29 cells were seeded in 24-well plates at a density of 2 × 105 cells per well and cultured for 24 h before subsequent studies. The cultured cells were incubated with NP, free Cur, CNP, SNP and CSNP for 48 h for the cells repairing the wound. Finally, in order to calculate the mobility, the quantization width in the case where the wound was observed through a microscope.
In vivo imaging
The female BALB/c nude mice were obtained from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All relevant experiments were carried out following the ethical rules enacted by Experimental Animal Ethics Committee in Beijing. CSNP and free IR780 were intravenously injected into tumor-bearing nude mice, and then the IVIS Spectrum in vivo optical imaging system (PerkinElmer, USA) and multispectral optical tomography system (MSOT invasion 128; iThera medical, Germany) were used for fluorescence and photoacoustic imaging at specific time points (1 h, 3 h, 6 h, 12 h, 24 h), respectively. All mice were immolated after imaging, and tumors and major organs were gathered for ex vivo fluorescence imaging further.
In vivo Antitumor Assessment
To establish a tumor model, 1×106 HT-29 cells dispersed in 100 µL of PBS were injected subcutaneously into the right hind limb of mice, which were divided into 5 groups (n = 5 per group) stochastically when the tumor volumes grown into approximately 150 mm3, and saline, free Cur, CNP, SNP and CSNP were injected through the tail vein (Cur 5 mg/kg, siCCAT1 2 mg/kg) every 2 days, respectively. The tumor volume and body weight of the experimental mice were measured every other day. After all mice were immolated on the 14th day, and tumors were excised and weighed.
Statistical analysis
All experiments were independently repeated at least 3 times (n = 3), unless otherwise stated. Data were expressed as mean ± SD. To evaluate the significance of the difference between the treatment groups student's t-test was used, and the statistical significance was *p < 0.05, **p < 0.01, *** p < 0.005 and ****p < 0.001.
{kind=link}