The present study identified a novel role for Psmb8 in regulating cardiomyocyte mitochondrial homeostasis during myocardial I/R injury in vivo and in vitro. Our results showed that Psmb8 overexpression in mice ameliorated I/R-induced cardiac dysfunction and injury associated with a reduction in mitochondrial fission by reducing Drp1 and increasing Mfn1/2 protein levels. Conversely, Psmb8-KO in mice had the opposite effects. These observations were verified in cultured cardiomyocytes infected with Ad-Psmb8 or siRNA-Psmb8 in vitro. Mechanistically, Psmb8 directly associates with Drp1 and enhances its degradation, which limits excessive mitochondrial fission, reactive oxygen species (ROS) production, ATP depletion, and subsequent cardiomyocyte apoptosis, thereby improving cardiac dysfunction. Therefore, our results suggest that Psmb8 is essential for maintaining cardiac mitochondrial dynamic balance by targeting Drp1 and highlight Psmb8 as a promising therapeutic target for cardiac I/R injury. A working model is presented in Fig. 9.
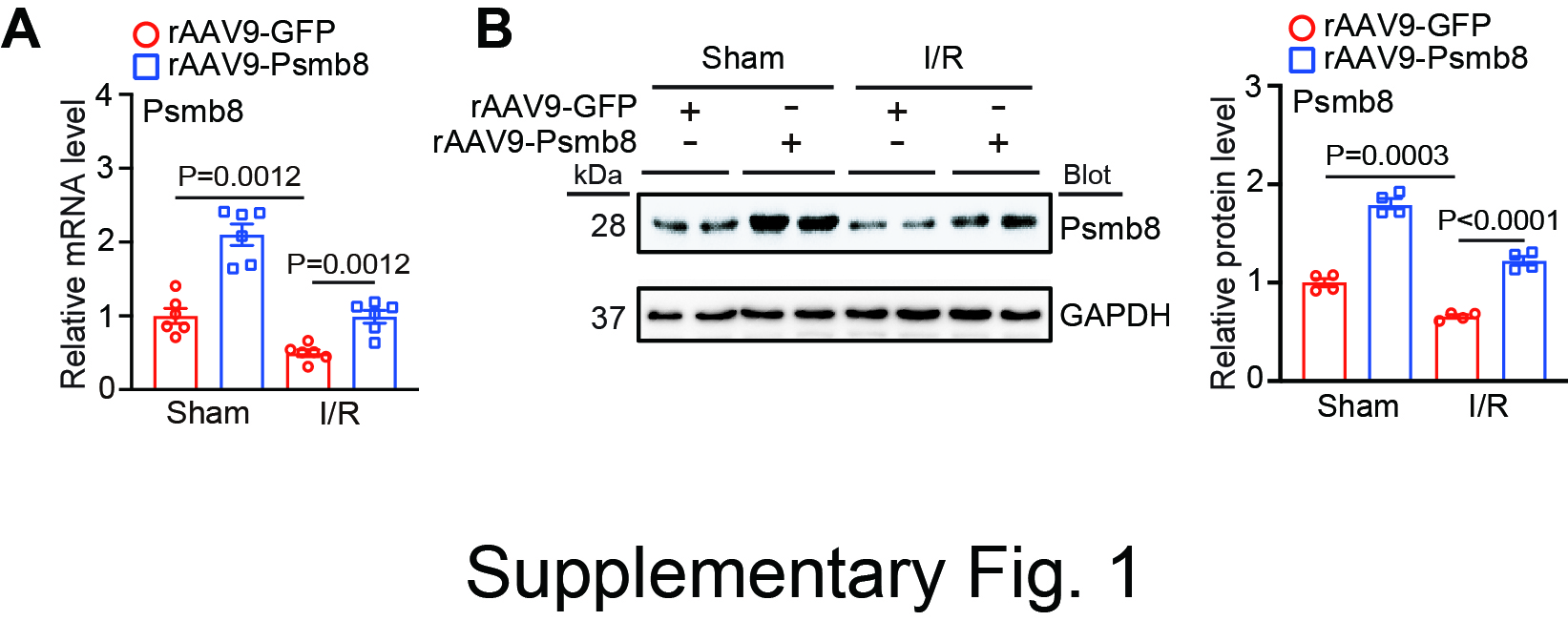
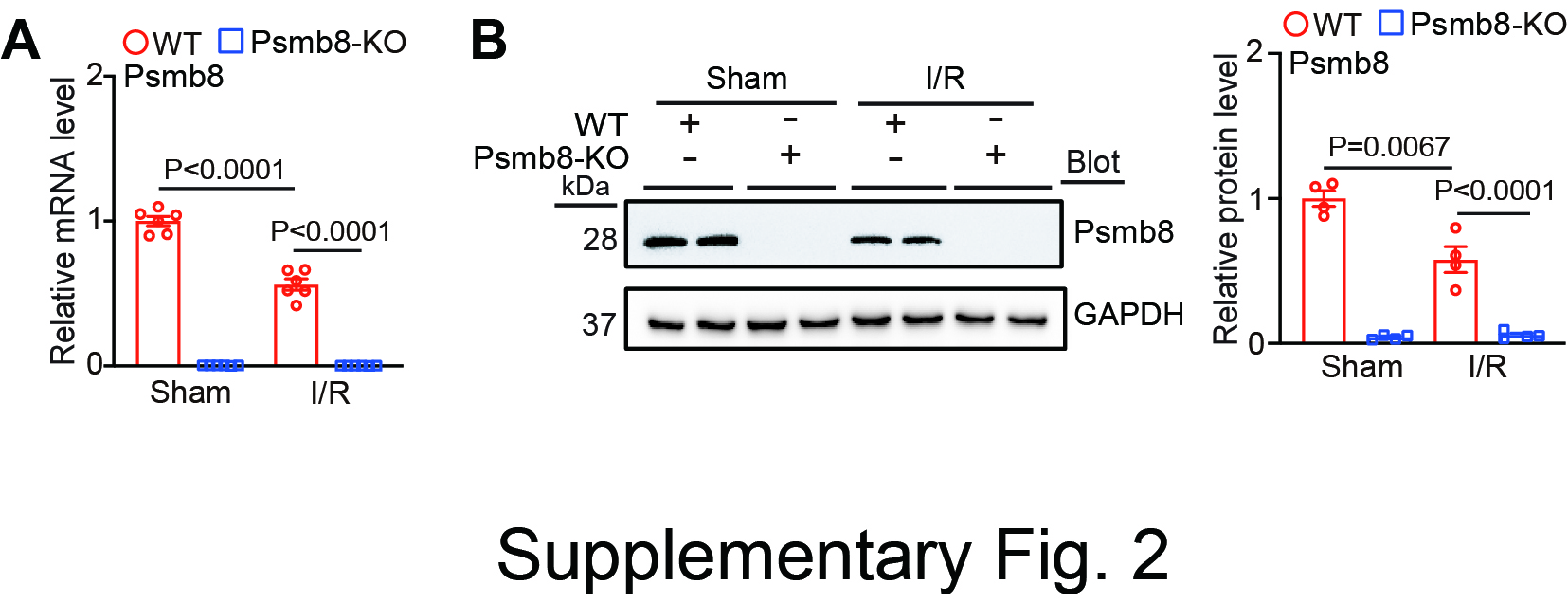
The immunoproteasome plays a central role in immunity and various inflammatory disorders. The expression levels of catalytic β immunosubunits, particularly β2i (Psmb10) and β5i (Psmb8), are dramatically upregulated in various tissues in mouse models in response to inflammatory and hypertensive stimuli and in human patients [5, 6]. Importantly, our recent studies indicate that the activation of Psmb10 and Psmb8 contributes to several cardiovascular diseases by regulating the stability of multiple protein substrates (ATG5, PTEN, ATRAP, IKBα, SOCS3, etc.) that are involved in autophagy, inflammation, oxidative stress, and apoptosis [7–12]. However, several studies have indicated that the expression levels and activities of the constitutive subunit (Psmb5) and immunosubunits (Psmb9 and Psmb10) are decreased in cardiac tissues and cells following ischaemic injury and are involved in cardiac and cerebral I/R injury [13, 17, 18]. For example, cardiomyocyte-specific overexpression of the β5-T60A mutation inhibits proteasome activity and dramatically exacerbates I/R-induced cardiac injury and dysfunction in mice, which is associated with increases in PTEN and PKC-δ protein levels and the inactivation of AKT [13]. Conversely, knockout of Psmb9 (β1i) abolishes ischaemic preconditioning-mediated cardioprotection against I/R injury by inhibiting PTEN degradation [14]. Furthermore, overexpression of PA28 (a proteasome activator) in cardiomyocytes prevents desmin-related cardiomyopathy and myocardial I/R injury [15]. More recently, we showed that cardiomyocyte overexpression of Psmb10 alleviated I/R-mediated cardiac infarction, apoptosis and dysfunction by targeting Parkin-Mfn1/2-mediated mitochondrial fusion [16]. However, the importance of another catalytic Psmb8 subunit in I/R-induced cardiac dysfunction has not been determined. Here, we extended the previous observations and further revealed that Psmb8 expression and activity were significantly reduced in mouse I/R-induced hearts and MI patients (Fig. 1). Overexpression of Psmb8 in cardiomyocytes significantly ameliorated I/R-induced cardiac dysfunction and damage, which were accompanied by decreased mitochondrial fission in mice (Figs. 2 and 3), and knockout of Psmb8 accelerated these effects (Fig. 4). The cardiomyocyte apoptosis and mitochondrial fission results in vivo were further confirmed in NRCMs infected with Ad-Psmb8 or siRNA-Psmb8 in vitro (Fig. 5). Thus, Psmb8 is involved in regulating mitochondrial fission and cardiac function during I/R injury.
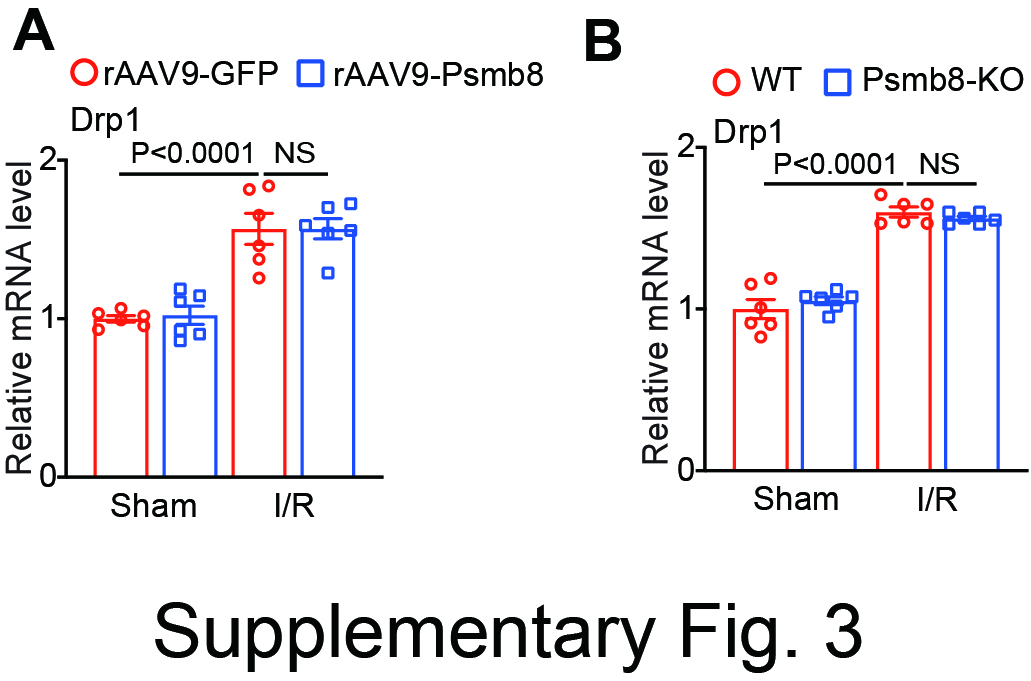
The mechanisms responsible for the pathogenesis of cardiac I/R injury are complex. The disruption of mitochondrial homeostasis is a central factor in the regulation of cardiac I/R damage [3, 30, 31]. Notably, Drp1 is a key fission protein that hydrolyses GTP to trigger mitochondrial fission, and this effect is facilitated by numerous adapter proteins, such as fission protein 1 (Fis1), mitochondrial fission factor (MFF), and mitochondrial dynamics proteins of 49 kDa and 51 kDa (MiD49 and MiD51, respectively), at the outer mitochondrial membrane (OMM) [25]. Moreover, Drp1-mediated mitochondrial fission is a major source of ROS, which play a pivotal role in cardiomyocyte death during I/R injury [3, 4]. Conversely, inhibition of Drp1 markedly reduces mitochondrial fission and apoptosis [25]. Thus, selective targeting of Drp1 may be a new therapeutic strategy for ischaemic disease. Multiple posttranslational modifications, including phosphorylation, SUMOylation, and ubiquitination, regulate Drp1 activity, translocation, and degradation [30]. Pathophysiological stimuli, such as ATP depletion, hypoxia and I/R injury, can trigger the translocation of Drp1 to the OMM through the phosphorylation/dephosphorylation of serine residues 616 and 637 [3]. Currently, several protein kinases, including CDK1, PKCδ, CaMKII, and PINK1, can phosphorylate Drp1 at serine 616 (Drp1S616), which leads to increased activity and subsequent mitochondrial fission [3, 32], while the Ca2+-dependent phosphatase calcineurin catalyses Drp1 dephosphorylation at serine 637 to inhibit mitochondrial fission, thereby ameliorating I/R-induced cardiac cell death and dysfunction [21, 33, 34]. Moreover, the stability of Drp1 is modulated by E3 ligase-dependent ubiquitination. Mitochondrial MARCH5 (MITOL) can promote Drp1 ubiquitination and proteasomal degradation and limit mitochondrial fission [26–28]. The E3 ligase Parkin also mediates Drp1 ubiquitination and degradation, resulting in an imbalance in mitochondrial dynamics in Parkinson’s disease [29], suggesting that ubiquitin modification regulates Drp1 stability and mitochondrial fission. Notably, Parkin protein levels are increased in I/R-induced hearts [16], which is consistent with the pattern of Drp1 expression observed in the present study (Fig. 3G, 4H), and exclude the involvement of Parkin in Drp1 degradation. Interestingly, our recent studies revealed that the I/R-induced upregulation of Drp1 expression is markedly reduced by several small molecule components, such as ursolic acid (UA), MK-886 and TCH-165, by increasing the expression and activity of immunosubunits (Psmb9, Psmb10 or Psmb8) [17, 18, 35]. However, the mechanism by which these immunosubunits enhance ubiquitinated Drp1 degradation has not been determined. Here, our data revealed a new mechanism by which Psmb8 directly interacted with Drp1 and enhanced its degradation (Fig. 6). Moreover, knockdown or inhibition of Drp1 in NRCMs robustly attenuated H/R-induced cardiomyocyte apoptosis and mitochondrial fission in Psmb8-deficient NRCMs (Fig. 5). The cardioprotective effect of Drp1 was further confirmed in I/R-treated Psmb8-KO mice after siDrp1 knockdown (Fig. 8), suggesting that Drp1 is a direct target of Psmb8 in cardiomyocytes. Thus, we identified a novel mechanism by which Psmb8 ameliorates cardiac I/R injury and dysfunction through improving the balance of mitochondrial dynamics by targeting Drp1 degradation.
To date, accumulating evidence has demonstrated that proteasomal activation is involved in different cardiovascular diseases induced by hypertension, angiotensin II infusion and pressure overload [7–11]. In contrast, deletion or inhibition of proteasome catalytic subunits (Psmb9, Psmb10 or Psmb5) and activators (PA28) are associated with exacerbation of cardiac I/R injury and cardiomyopathy [14, 15, 17, 18], suggesting that these factors could be novel targets for therapeutic intervention in hypertrophic or ischaemic heart diseases. The inhibition of proteasome activity with inhibitors such as bortezomib, epoxomicin, and PR-957 (ONX-0914) significantly attenuates hypertension, cardiac remodelling, heart failure, atrial fibrillation and retinopathy after angiotensin II infusion or pressure overload [7–11, 36, 37]. More recently, we identified several immunoproteasome inducers, including ursolic acid (UA), MK-886, TCH-165, and N-arachidonoylphenolamine (AM404) [38], which can protect against cardiac I/R damage by targeting the PP2A-AMPK and keap1-NRF2 signalling pathways, thereby leading to Drp1 degradation [17, 18, 35].
This study has several limitations. First, we did not determine the mechanisms by which I/R downregulates Psmb8 expression in cardiomyocytes; which specific E3 ligases promote Drp1 ubiquitination for degradation by Psmb8; how Psmb8 increases Mfn1 and Mfn2 expression in cardiomyocytes, or whether the activation of Psmb8 represents a therapeutic strategy for ischaemic heart disease in human patients. Moreover, our patient sample size was relatively small, and further large-scale multicentre studies are needed to verify our findings.
In summary, the present study revealed that Psmb8 expression was significantly decreased in I/R-induced hearts and H/R-induced cardiomyocytes, as was that in patients with MI. An increase in Psmb8 expression significantly attenuated I/R-mediated cardiac injury and dysfunction by targeting Drp1 for degradation and inhibiting excessive mitochondrial fission. Taken together, these findings provide new insights into the mechanism by which Psmb8 regulates Drp1 stability and subsequent cardiac I/R injury and suggest that activating Psmb8 may be a promising therapeutic strategy for ischaemic heart disease.
{kind=link}
{kind=link}
{kind=link}