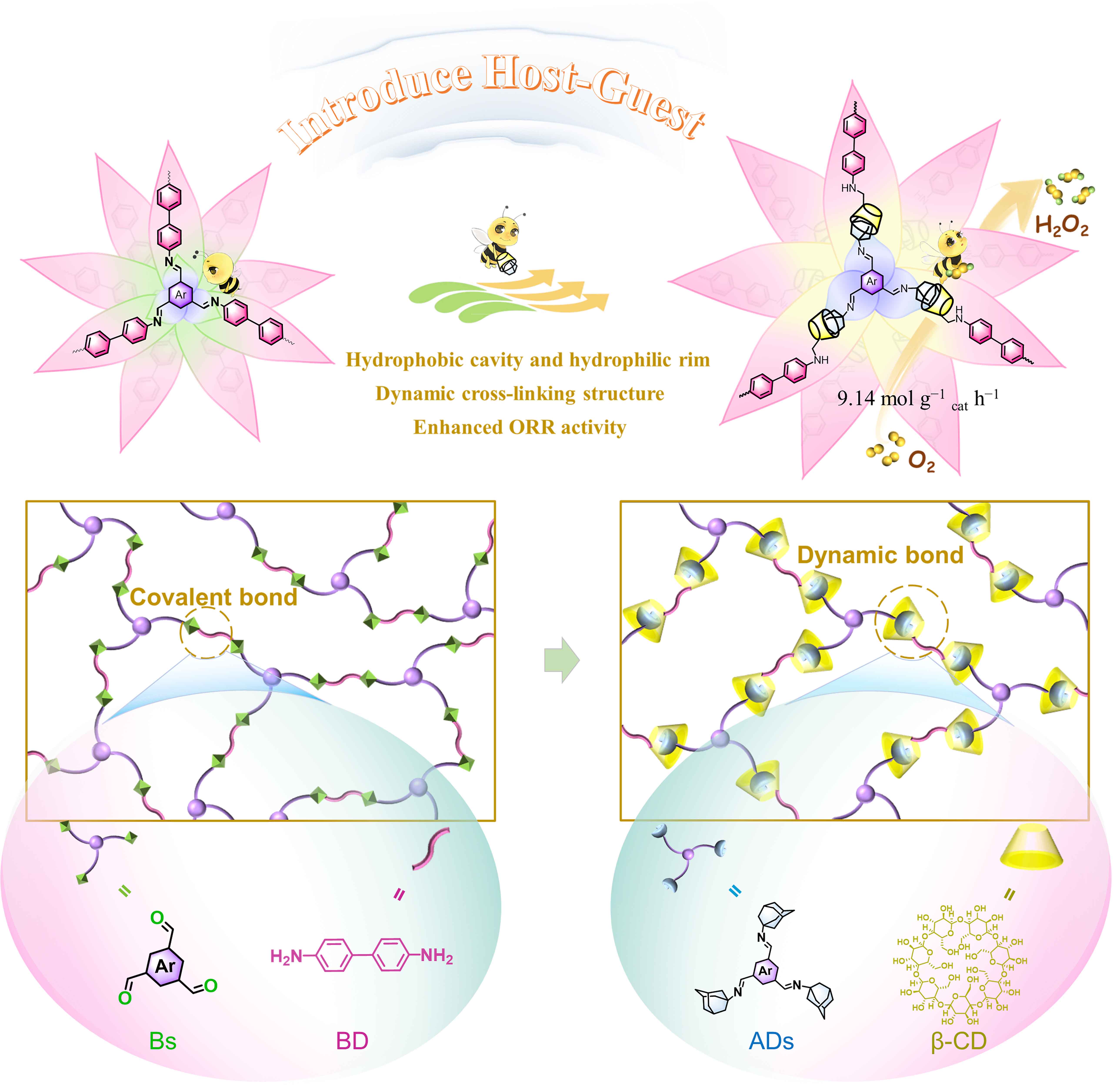
Synthesis of covalent-linked polymers and dynamic-crosslinked supramolecular polymers. To implement the dynamic and covalent bond strategy and intuitively investigate the impact of different bonding modes on catalytic activity, 4,4’-Biphenyldiamine (BD) as a planar linker, 1,3,5-Tris-(p-formylphenyl) benzene (TPB) and 1,3,5-Benzenetricarboxaldehyde (Ph) as linear building units were employed to directly synthesize two conjugated microporous scaffolders under improved solvothermal conditions, denoted as P-CN-TPB and P-CN-Ph. Two supramolecular polymers, named HG-CD-TPB and HG-CD-Ph, were further generated with the introduction of β-CD-adamantine noncovalent building blocks. Scheme 1 illustrates the use of "bee pollination" to promote "flowering maturity" by introducing host-guest inclusion structures into conjugated microporous frameworks. The hydrophobic cavity and hydrophilic rim features of β-CD enable the ability to promote the formation of a local electric field in the internal environment and accelerate the transfer of electrons, while the external hydrophilicity can also improve the electron transport rate. In addition, compared to covalent-linked polymers with analogous skeletons, the host-guest chemistry will endow supramolecular catalysts with dynamic cross-linking structures, enabling self-assembly and self-healing for efficient ORR activity.
The influence of different bonding modes on the electronic environment of metal-free organic molecules was further studied by density functional theory (DFT) calculation. The molecular orbital energy level, as a nonnegligible factor for electron distribution, has been extensively studied in electrocatalysis28. As schematically presented in Fig. 1, the host-guest-based HG-CD-TPB and HG-CD-Ph exhibit obvious D-A properties, contributing to charge transfer during the reaction. Quantitative molecular surface analysis was performed by combining electrostatic potential (ESP) and average local ionization energy (ALIE)29,30. In covalent bonded polymers, P-CN-TPB and P-CN-Ph, negative charges are concentrated on the electronegative N atoms, while after the introduction of β-CD-adamantine, most of the charges of HG-CD-TPB and HG-CD-Ph are attracted by the interaction between host and guest forces, breaking the relatively uniform charge distribution within the molecules, which is conducive to the improvement of catalytic activity31,32. Additionally, the minimum ALIE values of both supramolecules and polymers are located proximal to the C = N(N), indicating a weaker charge binding force, a stronger electron activity, and an easier occurrence of electrophilic reaction in this region, thereby favoring 2e− ORR33,34.
To analyze the chemical structures of the as-prepared polymers, Fourier transform infrared (FT-IR) spectroscopy was conducted (Fig. 2a). The FT-IR spectra of carbon-based P-CN-Ph and P-CN-TPB with covalent bonds exhibited the characteristic C = N stretching vibration mode at 1625 cm− 1, accompanied by the absence of the N–H stretching bands at 3323 and 3393 cm− 1 from the diamine monomers as well as the disappearance of C = O stretching bands of these monomers at 1690 cm− 1 (Figures S1 − 2). The FT-IR data provides evidence for successful imine condensation between aldehyde and amino monomers35. The conductivity was further assessed through current-voltage (I-V) tests (Fig. 2b). As can be seen, dynamic-crosslinked supramolecular polymers exhibited higher conductivities compared to covalent-linked microporous polymers. The electronic structure of these electrocatalysts was evaluated using solid ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) (Fig. 2c). In comparison to the covalent bonded P-CN-Ph and P-CN-TPB, the host-guest-based HG-CD-Ph and HG-CD-TPB with smaller band gaps of 1.67 and 2.00 eV, respectively, displayed distinct red-shifted absorption (Figure S3). Moreover, thermal gravimetric analysis (TGA) and derivative thermogravimetric (DTG) analysis demonstrate the good thermal stability of both synthesized host-guest and covalent bond catalysts (Fig. 2d). The ORR is widely acknowledged as a three-phase interface reaction involving gas, liquid, and solid phases36.Therefore, the wettability characteristics of dynamic-crosslinked supramolecular polymers and covalent-linked polymers were investigated to gain insights into their hydrophobicity/hydrophilicity (Fig. 2e). It should be noted that the variation in connected bonds significantly influences the hydrophobic or hydrophilic nature of these electrocatalysts, thereby modulating the interaction between oxygen-active species and active sites within their frameworks. The amorphous morphology of dynamic-crosslinked supramolecular polymers and covalent-linked polymers was confirmed through Powder X-ray diffraction (PXRD) measurements (Figure S4). Furthermore, compared with the PXRD of supramolecular precursors, the disappearance of small molecular peaks in dynamic-crosslinked supramolecular polymers provides preliminary evidence for the formation of inclusion complexes between adamantane and β-CD, as well as indicating a host-guest interaction (Figures S5 − 6). In addition, the FT-IR shift of O − H bonds can further prove that the host-guest interaction is successfully generated (Figures S7 − 8)37. Two-dimensional nuclear Overhauser effect spectroscopy (2D NOESY) NMR is an efficient and sensitive method for the analysis of near proton interaction. Therefore, it was further employed to precisely confirm the host-guest interaction between adamantane and β-CD. Figures 2f and S9 display the correlation between the − CH2 protons of adamantanes and the protons (C(3)H and C(5)H) in the β-CD cavity, represented by a green rectangular signal, validating successful self-assembly of the host β-CD and the guest adamantanes38,39.
Scanning electron microscopy (SEM) showed stacked spheres of covalent-linked polymers and porous masses of dynamic-crosslinked supramolecular polymers (Fig. 3a–d). Specific observations demonstrated that P-CN-TPB exhibits a wire globule-covered sphere, and P-CN-Ph features a globule-filled sphere, whereas, a microporous multi-layer shell of HG-CD-TPB and HG-CD-Ph presents a large porous random cube (Fig. 3e-h), indicating that modifications in the connected bonds resulting from host-guest strategy can significantly affect molecule growth patterns. To gain further insights into the morphology of covalent-linked polymers and dynamic-crosslinked supramolecular polymers, energy-dispersive X-ray spectroscopy (EDS) mapping photos and transmission electron microscopy (TEM) were conducted, confirming the uniform distribution of characteristic elements throughout these materials (Fig. 3i − l).
Electrocatalytic 2e−-ORR characterizations. To investigate the ORHP properties of these supramolecular catalysts, a rotating ring-disc electrode (RRDE) was used to perform electrochemical tests in an aqueous solution (O2-saturated 0.1 M KOH electrolyte). As depicted in Figs. 4a–c and S10–13, the host-guest-based polymers (HG-CD-Ph and HG-CD-TPB) exhibit higher ring current and activity in ORHP compared to the covalent bonded polymers (P-CN-Ph and P-CN-TPB), and even superior to the recently reported most covalent bonded catalysts (Figs. 4h and S14, and Table S1), particularly the optimum selectivity for H2O2 of HG-CD-Ph (94.93% at 0.4 V vs. RHE). The electron-transfer number of HG-CD-Ph also decreases to 2.10 at 0.4 V, showing an obvious two-electron transfer process. A similar phenomenon is also found in neutral electrolytes (Figure S15). Moreover, electrochemical tests conducted on supramolecular precursors revealed significantly diminished properties compared to those of supramolecules HG-CD-TPB and HG-CD-Ph, indicating that the dynamic bond formed through host-guest interaction can effectively regulate the reaction process and enhance the catalytic efficiency (Figures S16–17). The disparity in performance between dynamic-crosslinked supramolecular polymers and covalent-linked polymers is ascribed to the different connected bonds as well as hydrophilic and hydrophobic characteristics, implying that dynamic "dual-channel" regulation induced by host-guest strategy plays a pivotal role in enhancing electrocatalytic activities40,41. HG-CD-Ph and HG-CD-TPB have smaller Tafel slopes of 75.56 and 76.33 mV dec− 1, respectively, compared with those of P-CN-Ph and P-CN-TPB (77.29 mV dec− 1 and 79.81 mV dec− 1), demonstrating the faster charge-transfer kinetics of dynamic-crosslinked supramolecular polymers under alkaline conditions (Fig. 4d). The electrochemically active surface areas (ECSAs) were estimated using the electrochemical double-layer capacitance (Cdl) (Figs. 4e and S18–22). The results confirmed that the central phenyl group-containing HG-CD-Ph exhibited the highest Cdl value, clearly indicating its superior active density. Furthermore, the smaller curve radius of dynamic-crosslinked supramolecular polymers, as measured by electrochemical impedance spectroscopy (EIS), demonstrated that the host-guest induced D–A structure was conducive to a reduced charge transfer resistance (Rct) (Fig. 4f)42. In addition, the long-term stability of HG-CD-Ph was assessed through a 10-hour test in an O2-saturated 0.1 M KOH electrolyte (Fig. 4g). The result shows that HG-CD-Ph maintains excellent stability for prolonged H2O2 production, with a remarkable activity retention rate of 94% over the course of 10 hours at 0.4 V under alkaline conditions.
Motivated by the excellent 2e− ORR performance observed in RRDE, the H2O2 electrosynthesis was investigated on a practical three-phase flow cell using chronopotentiometry (Fig. 5a). After 1 hour of electrolysis, the H2O2 concentration in the cathodic tank’s electrolyte was analyzed to calculate the H2O2 production rate and Faradic efficiency (FE). To determine the H2O2 concentration, traditional Ce(SO4)2 titration and a well-fitted calibration curve for UV-vis spectrophotometric determination of Ce4+ in an aqueous solution were performed as shown in Figures S23 − 24. The yield and FE at different constant currents revealed that HG-CD-Ph exhibited the highest FE value at 80 mA cm− 2 (Fig. 5b). Therefore, the yields of different samples at 80 mA cm− 2 were selected for comparison (Figures S25–28). The H2O2 yield of HG-CD-Ph, as shown in Fig. 5c-d, is remarkably high at 9.14 mol g− 1 cat h− 1 (1462.75 µmol h− 1 with FE of 98.01%), surpassing the performance of HG-CD-TPB, P-CN-Ph, and P-CN-TPB under the test current. The Fenton reaction is a well-established method for pollutant treatment, wherein the reaction between Fe2+ and H2O2 generates hydroxyl radicals that can effectively react with pollutants43. Leveraging the exceptional 2e− ORR electrocatalytic activity of the central phenyl group-containing HG-CD-Ph, we further used the generated H2O2 to degrade organic dyes (malachite green, rhodamine B, and methylene blue), (the detailed experiment was provided in Supporting Information). The optical image clearly illustrates the rapid fading of malachite green, rhodamine B, and methylene blue from colored to colorless within a few minutes (Figs. 5e–f and S29). By providing additional evidence with experiments on organic dye degradation, this study further supports HG-CD-Ph's potential as an electrocatalyst for efficient H2O2 production. The results of all aforementioned studies consistently indicate that HG-CD-Ph exhibits superior activity in the production of H2O2 (Fig. 5g).
Theoretical calculations. To gain further insights into the distinct performance of P-CN-TPB, P-CN-Ph, HG-CD-TPB, and HG-CD-Ph in 2e− ORR, DFT calculations were carried out for these four polymer models. All structures along the potential energy surfaces were optimized without any constraints. Frequency analysis was performed to confirm that all optimized geometries corresponded to local minima and to obtain the Gibbs free energies44–46. In the calculation, the nomenclature of P-CN-TPB, P-CN-Ph, HG-CD-TPB, and HG-CD-Ph for these model molecules was maintained (Fig. 6a). In addition, three possible active sites were selected to determine the overpotentials for H2O2 production (Figures S30–33 and Tables S2–5). As shown in Fig. 6b, we presented key geometrical parameters and the Mulliken charges on main atoms at stationary points along the reaction pathway of the four polymers catalyzing 2e− ORR47,48. The shortened C–O bond results in a stronger charge transfer between HG-CD-Ph and O2. Figure 6c illustrates the partitional density of states (PDOS) for total (purple lines), C atoms (green lines), N atoms (orange lines), and O atoms (pink lines) on P-CN-TPB, P-CN-Ph, HG-CD-TPB, and HG-CD-Ph. It could be found that the contribution of C is larger in host-guest-based molecules HG-CD-TPB and HG-CD-Ph than in covalent-bond-based polymers P-CN-TPB and P-CN-Ph, indicating that the introduction of host-guest strengthens the electronic density of C atoms. This enhancement may facilitate the interaction between the C and O2 moieties during the reaction49. All above-calculated results are consistent with the experimental observations.
To gain a comprehensive understanding of the catalytic performance of dynamic-bond-based HG-CD-Ph and covalent-bond-based P-CN-Ph, the actual catalytically active centers were investigated by combining the in situ attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) spectroscopic characterization and DFT calculations. Two theoretical transmission models (Figs. 7a and S34) were constructed to elucidate the potential transmission locations of OOH* intermediate. ATR-FTIR spectroscopic tests were conducted to detect the crucially adsorbed OOH* on HG-CD-Ph and P-CN-Ph during the electrolytic H2O2 synthesis (Figs. 7b and S35). A weak transmission band at about 1210 cm− 1 appears at a potential of 0.7 V vs RHE, which is gradually enhanced by decreasing the potential (Fig. 7b). These transmission bands on HG-CD-PH can be assigned to O–O stretching vibration of OOH*, slightly shifted towards lower wavenumbers compared to those obtained from DFT calculations likely due to different transmission sites. Additionally, the bands at 976 cm− 1 that increase with negatively shifted potential can be reasonably assigned to the C–O stretching mode of OOH*. Furthermore, the spectral band assigned to adsorbed hydroperoxides (usually at 1386 cm− 1) is relatively poorer, as the generated H2O2 is immediately converted into HO2− products in alkaline environments50–52. Overall, the detection of potential-dependent transmitted hydroperoxy bands provides evidence for the OOH* mediated two-electron ORR pathway on both HG-CD-Ph and P-CN-Ph catalysts. To further ensure the catalytic active site of the as-prepared materials, additional DFT calculations were performed on the model compounds. The electron localization function (ELF) reveals that HG-CD-TPB and HG-CD-Ph enhanced localization around the C = N(N) atoms region (Fig. 7c, Left), conducting the ORR catalytic reaction53,54. Electrophilic sites were further predicted by Fukui function to identify active sites. It is worth noteworthy that the isosurface maps progressively increase from P-CN-TPB, P-CN-Ph, and HG-CD-TPB to HG-CD-Ph. The highest distribution of isosurface map in HG-CD-Ph will favorably contribute to the adsorption of electrophilic oxygen intermediates (Fig. 7c, Right)55,56. The overpotential of the C atom on the adamantane (site–1) for the central phenyl group-containing HG-CD-Ph (0.07 eV), which is influenced by host-guest interaction, was lower than that of P-CN-Ph (0.5 eV) (Fig. 7d). Obviously, the dynamic bonds promote the positive ORR activity. To further explore the impact of connected bonds on activity and selectivity, the electron transfer from HG-CD-Ph to O2 was examined. Figure 7e shows the highest occupied molecular orbital (HOMO) diagram of various polymers. The HOMO energy of HG-CD-Ph is − 4.88 eV, closely resembling the LUMO energy of oxygen, resulting in easier electron transfer from HG-CD-Ph to oxygen57. Therefore, the easier adsorption of oxygen during ORR elucidates the heightened catalytic activity of HG-CD-Ph.
The reported studies have demonstrated that the ORR activity and selectivity can be attributed to the binding free energy of reaction intermediate and the ORHP process58. Consequently, the catalytic activity can be determined by the corresponding energy level of the crucial intermediate (OOH*). The ΔGOOH* (binding energy of OOH*) was employed as a descriptor and construct an activity volcano to estimate the activities of different connected bonds. The maximum UL (zero overpotential at the top of the volcano) is determined to be 0.70 V. The calculated UL as a function of ΔGOOH* for the ORHP process of these catalysts is shown in Fig. 7f. The structures positioned on the right side of the volcano present the weak binding energy of OOH* and on the left side they strongly bind OOH*, while the middle represents the theoretical optimal ΔGOOH*59,60. As a result, variations in binding strength of OOH* in dynamic-crosslinked supramolecular polymers and covalent-linked polymers lead to diverse ORHP activities. The sites–1 on HG-CD-Ph is closer to the top of the volcano map, demonstrating the host-guest decoration is beneficial to ORHP for more moderate binding of OOH*.
{kind=link}
{kind=link}