2.1. Materials and animals
N-Isopropylacrylamide (NIPAM) and acrylic acid (AA) were procured from Tokyo, Japan. Poppy iodine oil was obtained from Jiangsu Hengrui Pharmaceutical Co., Ltd. Medical three-way tubes were sourced from Yangzhou Yangsheng Pharmaceutical Technology Co., Ltd. N, N'-methylene bisacrylamide (MBA), octadecyl trimethyl ammonium chloride (STAC), 2,2'-azobis(2-methylpropionamidine) dihydrochloride (AAPH), and rhodamine-B were purchased from Aladdin Chemical Reagent Co., Ltd. 4% paraformaldehyde and pentobarbital sodium were acquired from McLean Reagent Co., Ltd. Lidocaine hydrochloride and gentamicin sulfate were obtained from Huabei Pharmaceutical Co., Ltd. Heparin sodium injection was sourced from Changzhou Qianhong Biochemical Pharmaceutical Co., Ltd. Sodium chloride injection was purchased from Wuhan Binhu Shuanghe Pharmaceutical Co., Ltd. Sodium lauryl sulfate (SDS) and ethylene glycol-bis-(2-aminoethyl) tetraacetic acid (EGTA) were procured from Aladdin Biochemical Technology Co., Ltd. Milli-Q ultrapure water (18.2 MΩ) was utilized for experimental verification. DNA reagent extraction kits were obtained from QIAGEN.
Adult New Zealand white rabbits (males, weighing 2.5–3.0 kg) and Sprague Dawley rats (males, weighing 300–400 g) were obtained from the Laboratory Animal Center of Hubei University of Science and Technology. All animal experiments adhered to the Guide for the Care and Use of Laboratory Animals issued by the Hubei Provincial Department of Science and Technology and were approved by the Animal Ethics Committee of Hubei University of Science and Technology (approval number: 2024-02-002).
2.2. Synthesis of PNAs
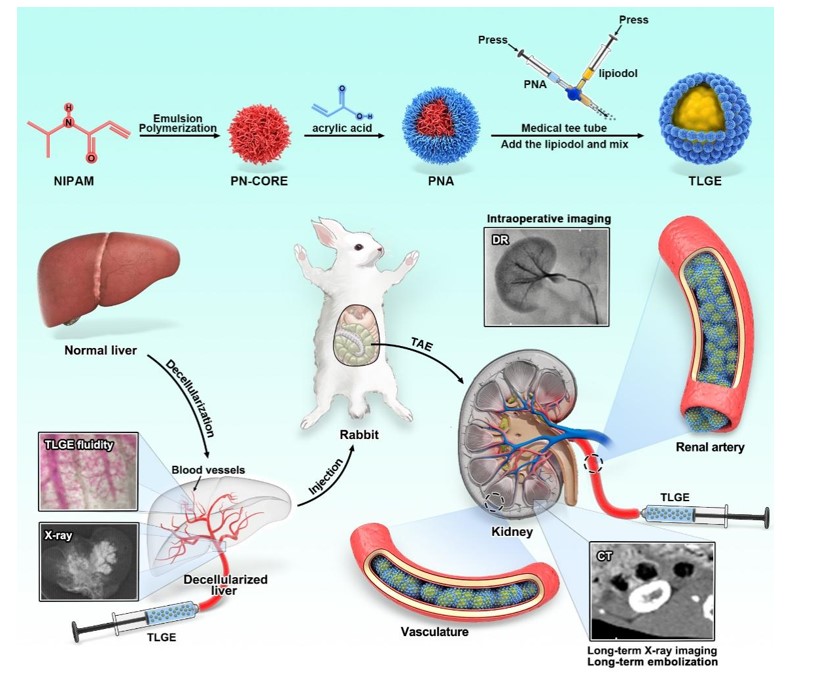
First, the formula was designed by emulsion polymerization, and the optimal formula ratio for the preparation of the hydrogel was determined. Briefly, MBA (0.03 g, 0.19 mmol), STAC (0.05 g, 0.29 mmol), NIPAM (1.7 g, 15 mmol), and AAPH (0.08 mg, 0.14 mmol) were dissolved in 150 mL of ultrapure water, added to a three-necked flask, and stirred in an oil bath at 80°C and 250 rpm until the solution turned blue, followed by the addition of AA (215 L, 3 mmol). After the solution changed from light blue to milky white, the reaction was continued for 4 h. The reaction-completed gel solution was dialyzed against mobile water using a Molecular Weight Cut-Off Point (14,000 Da) for 3 days. On completion of dialysis, the samples were freeze-dried in a -80°C refrigerator for 4 h and then freeze-dried for 2–3 days until powdered ready for use.
2.3. Preparation of TLGE
A mixture of 6 wt.% and 8 wt.% PNAs and lipiodol was prepared for blending. Lipiodol volumes of 0.1 mL, 0.3 mL, 0.5 mL, 0.7 mL, and 0.9 mL were chosen, while corresponding volumes of 6 wt.% and 8 wt.% PNAs were added to yield a total volume of 1 mL. This ensured lipiodol concentration ratios of 10%, 30%, 50%, 70%, and 90%. The combined solutions were transferred into a 1 mL syringe connected to a medical three-way tube and alternately pushed and mixed for 100 cycles to create TLGE, which was then transferred to a 1 mL glass bottle for storage. Due to excessive lipiodol content, the 70% and 90% lipiodol blends did not gel and were discarded. Therefore, experiments comparing 10%, 30%, and 50% lipiodol mixed with 6 wt.% and 8 wt.% PNAs by weight were conducted.
2.4. Characterization of the PNAs
The PNAs were diluted to 1 wt.% by content, heated at 1°C/min, and the change in transmittance at 500 nm was measured using an ultraviolet spectrophotometer (Beijing General Analytical Instruments, China) from 25°C to 40°C. The morphology of the PNA nanogels was observed by transmission electron microscopy at 200 kV (Tecnai G2 20 FEI Corp, Netherlands). The nanogel aqueous dispersion was diluted with ultrapure water to 1 mg/mL. After 10 min of ultrasonication, 10 µL of the suspension was dropped on a TEM copper net coated with carbon film (300 mesh) at 25°C, dried, and then stained with phosphotungstic acid solution (0.1 wt.%) for observation.
The nanogel was diluted to 0.2 wt.% in ultrapure water, and 2 mL was removed and placed into a particle size sample pool for testing. The temperatures were set at 25°C and 37°C. A helium-neon laser (λ = 633 nm) was used as the light source, and the laser was balanced at a 90° scattering angle for 200 s before being circulated four times. Particle size analysis was conducted using a dynamic light scattering particle size analyzer, which produced a particle size distribution curve. Following this, 2 mL of gel dispersion liquid was transferred into the sample cell, with careful removal of bubbles, and then subjected to Zeta potential measurement using a dynamic scattering particle size analyzer.
2.5 Characterization of the TLGE emulsion
Lipiodol concentrations of 10%, 30%, and 50% were combined with PNAs at 6 wt.% and 8 wt.% content. Subsequently, 1 mL of the mixture was placed into a clear glass bottle. The bottle was then kept at room temperature (25°C) and photographed continuously for 1 h, 2 h, 3 h, 1 day, 3 days, and 10 days to evaluate its in vitro stability, oil phase separation, and emulsion migration. This procedure aimed to determine the optimal formulation ratio.
Following the mixing of 10%, 30%, and 50% lipiodol with 6 wt.% PNAs, the resulting emulsions were transferred into 2 mL glass bottles. Subsequently, the emulsions were photographed using a fluorescence electron microscope at room temperature (25°C) to examine their morphology.
The PNAs nanogel was stained with rhodamine-B and then mixed with 50% lipiodol for an in vitro injection experiment. Morphological changes were observed at both 25°C and 37°C. Various types of catheters were utilized for injection to investigate the impact of TLGE on vascular casting.
TLGE was subjected to CT scanning to observe its developability and corresponding CT value. The rheological properties of TLGE and PNAs nanogels were analyzed using a high-speed rotating rheometer (Anton Paar MCR92, Austria) under the following test conditions: parallel plates (PP50, Φ = 40 mm) with a gap of 0.5 mm, stress of 0.5 Pa, heating rate of 2.0°C/min, and frequency of 1.0 Hz. The G' and G" moduli of the PNAs and TLGE were determined over the temperature range of 25°C to 40°C. The viscosities of the PNA and TLGE nanogels were measured at 25°C, and the shear rates ranged from 150 s–1 to 2580 s–1.
2.6 Evaluation of the ex vivo live model
Twenty male SD rats, aged 8–10 weeks and weighing 300–400 g, were selected with SPF grade. They were housed under standard conditions with a 12-h light-dark cycle, having unrestricted access to water, and kept at a temperature of 22–25°C with a relative humidity of 50–60%. Our investigation into the liquid fluidity and imaging of TLGE utilized an ex vivo model of an acellular liver. The livers of male Sprague Dawley rats were euthanized by carbon dioxide asphyxia, and the whole liver was removed after being rinsed with 20 mL of PBS through the portal vein and then quickly placed at -80°C for 24 h. After 24 h, the liver was removed and slowly thawed at room temperature. Following this, a syringe pump (Mindray BeneFusion VP3 infusion pump) was employed to administer normal saline at a rate of 120 drops/min, along with 0.25% EGTA for 3 h, and 1% SDS for 6 h into the portal vein. Both the normal liver and the decellularized liver were subsequently fixed in 4% paraformaldehyde for 24 h, dehydrated using a tissue gradient ethanol, cleared with chloroform, and processed into liver specimens using the standard paraffin embedding technique. After the cells were removed, the sections were cut at a thickness of 4 µm. Hematoxylin and eosin (H&E) staining was used for histological analysis. For comparison with the decellularized liver, the nuclei and surrounding tissues were observed. Normal liver and acellular liver specimens were selected for immunofluorescence evaluation using rabbit polyclonal anti-type I collagen antibodies (Abcam, ab254349), rabbit polyclonal anti-laminin antibodies (Abcam, ab11575), and rabbit polyclonal anti-fibronectin antibodies (Servicebio, GB13091). Additionally, the liver morphology was examined using a scanning electron microscope (SU8100). Normal and decellularized liver tissue will be fixed with 2.5% glutaraldehyde for 24 h, dehydrated in an alcohol solution for 15–20 min, frozen at − 80°C for 2 h, and freeze-dried for 24 h. Normal and decellularized liver tissue will be sprayed with a platinum layer sputtering at 10 nm, after which scanning electron microscopy (SEM) images will be acquired at an accelerating voltage of 3 kV. A digital medical X-ray radiography system (DP528-B) was used at 55 kV, a current of 320.0 mA, a time of 12.00 ms, an EI of 57338, and 161.86 µGy. The acellular liver underwent imaging using a digital medical X-ray system. Lipiodol was then injected into the liver through the portal vein, and immediate imaging was conducted using DR. The decellularized liver was observed using an SZ680 stereoscopic microscope, with adjustments made to position and focal length. Intravenous injection of TLGE through the portal vein was performed at a rate of 0.5 mL/s, with vessel access observed at 1 s, 2 s, 3 s, 4 s, and 5 s intervals. DNA content analysis was carried out on six rats, comparing acellular and normal livers. After 10 h of irrigation, the decellularized liver underwent freeze-drying using a vacuum freeze-drying machine, alongside the normal liver. DNA extraction from both types of liver followed the kit's instructions, and the DNA concentration and purity were assessed using Nanodrop 2000. Agarose (0.75 g) was weighed into a conical bottle, and 50 mL of buffer solution was added. The samples were then microwaved and boiled until the agarose melted. The mixture was shaken well to form a 1.5% agarose gel. An equal volume of loading buffer was added to the sample (3–5 µL), and then the sample was added to the small tank of the rubber plate with a 10 µL micropipette. After the sample was added, the gel plate was immediately subjected to electrophoresis at 170 V, and the sample was moved from the negative electrode (black) to the positive electrode (red). After electrophoresis, the gel was removed, and the gel was observed under an ultraviolet lamp. The fluorescence bands were displayed in the presence of DNA, and a gel imaging system was used to take and save the images.
2.7 Application value of TLGE in renal artery embolization in normal rabbits
Embolization experiments were carried out in New Zealand rabbits involving their normal right kidneys. Twelve adult rabbits (males, weighing 3.0–3.5 kg) were specifically chosen for the study. Feeding conditions entailed maintaining temperatures between 25–27°C, humidity levels at 50–60%, ventilation frequencies ranging from 10–15 times/h, an air flow rate of 0.1 m/s, colony counts of ≤ 30/dish, noise levels below 50 dB, and 12-h illumination periods during both day and night. Feeding sessions were conducted three times daily.
Normal New Zealand rabbits were selected and euthanized immediately after 42 days of renal embolization with TLGE. Following a two-week adaptation period and a 12-h fast, pentobarbital sodium (30 mg/kg) was administered intravenously through the rabbit ears. Anesthesia took effect 15 min later, immobilizing them in a supine position. The inguinal fur was shaved and disinfected, and the femoral artery was separated using eye forceps after local anesthesia with lidocaine. Inserting an 18 g needle into the proximal artery along with a 4f coaxial microcatheter (Terumo, Tokyo, Japan), renal arteries underwent arterial angiography with iodixanol (300 mgI/mL, 0.5 mL/s) to confirm vessel patency. TLGE was injected into the rabbit renal artery, and real-time observation of emboli entering the renal artery at 1, 3, 5, and 7 s post-injection was performed using Digital Subtraction Angiography (DSA, Siemens BicorTop, Germany). Follow-up assessments were conducted seven days, 21 days, and 42 days post-embolization using Somatom Sensing 64 Slice (Siemens, Germany) with ioxatriol (300 mgI/mL–1) as the contrast agent. Scanning parameters were set at tube voltage: 80 kV and tube current: 60 mA. Renal artery embolization and vascular recanalization were monitored. After 42 days of daily treatment with intramuscular injection of 1–20 000 IU penicillin sodium, administered three days after surgery, rabbits were euthanized. Liver, spleen, lung, and kidney tissues were processed with H&E staining and immersed in 4% paraformaldehyde for 24 h. The tissues were then embedded in paraffin and sectioned. The sections were hydrated with graded ethanol and stained with H&E to analyze pathological changes after embolization. The left and right kidneys were removed and sectioned along the longitudinal axis for analysis of differences.
2.8 Evaluation of Biocompatibility
Blood (2 mL) from New Zealand rabbits was combined with normal saline (1,500 rpm × 10 min), and centrifuged (1,500 rpm × 10 min) until the supernatant cleared, yielding a 2% suspension of red blood cells. The experimental group consisted of TLGE emulsion containing 6 wt.% PNAs mixed with 50% lipiodol, diluted to concentrations of 5 mg/mL, 2.5 mg/mL, 1.25 mg/mL, and 0.625 mg/mL. Normal saline served as the negative control, while ultra-pure water acted as the positive control for treatment.
Five blood compatibility experiments were conducted in each group. In each group, 300 µL of solution was added to a 1.5 mL EP tube, along with 300 µL of red blood cell suspension. After incubation at a constant temperature for 1 h, the samples were centrifuged at 3000 rpm for 25 min, and 100 µL of supernatant was transferred into a 96-well plate. The hemolysis rate (Hr%) was calculated using a microplate reader (λ = 540 nm, 1420 MultiLabel Counter, Perkin Elmer).
$$Hr\%=\frac{{OD}_{s}-{OD}_{n}}{{OD}_{n}-{OD}_{b}}\times 100\%$$
ODs represent the absorbance of the experimental group, ODn represents the absorbance of the negative control group, and ODb represents the absorbance of the blank group.
The MTT method was employed to assess the cytotoxicity of HepG2 cells. Once the HepG2 cells reached the logarithmic growth phase, they were harvested following trypsin digestion. The cells were diluted to 1 × 105 cells/mL in DMEM and incubated for 24 h in 96-well plates filled with 4–5 × 103 cells/well. The TLGE emulsions of 6 wt.% PNAs mixed with 50% lipiodol and PNAs at different concentrations were diluted to 0.4 mg/mL, 0.2 mg/mL, 0.1 mg/mL, 0.05 mg/mL, and 0.025 mg/mL in DMEM. After that, the old medium was discarded. One hundred microliter samples of different concentrations were added to a 96-well plate and incubated with HepG2 cells for 24 h. DMEM containing cells was used as the negative control, and DMEM without cells was used as the blank control. Then, 50 mg of MTT powder and 10 mL of PBS were added, and the final concentration was 5 mg/mL. Then, 20 µL of 5 mg/mL MTT solution was added to each well, the plate was cultured in the dark for 4 h, the supernatant was discarded, 150 µL of DMSO solution was added to each well, the plate was shaken with a shaker at a constant temperature for 10 min, and the absorbance was measured with an automatic enzyme-labeled reader at 570 nm. The cell viability was calculated according to the following formula:
$$Cv\%= \frac{{OD}_{s}-{OD}_{b}}{{OD}_{n}-{OD}_{b}}\times 100\%$$
ODs, ODb, and ODn represent the absorbance values of the sample group, blank group, and negative control group, respectively.
{kind=link}