Structure and composition of the M. pauhoi mitochondrial genome
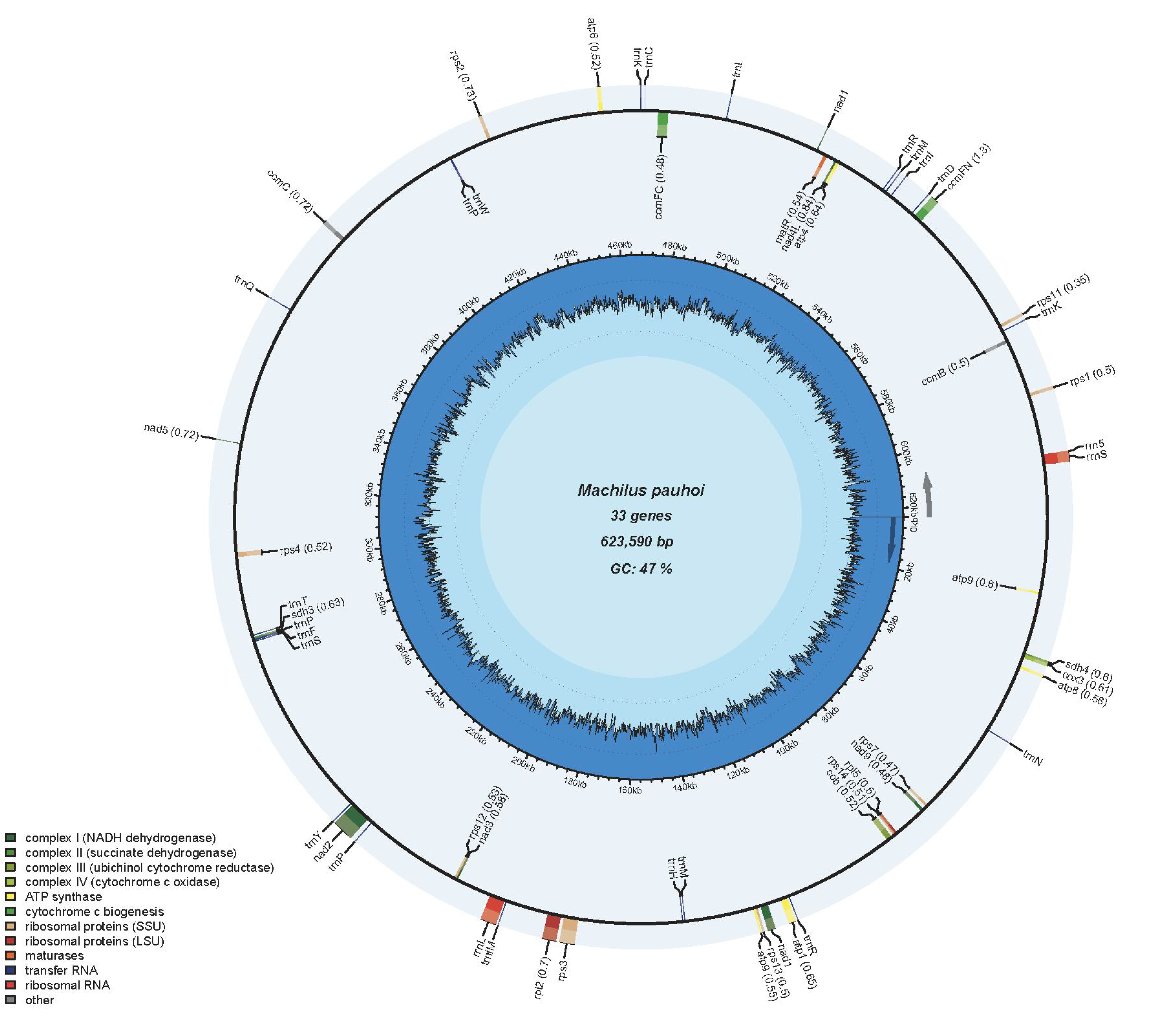
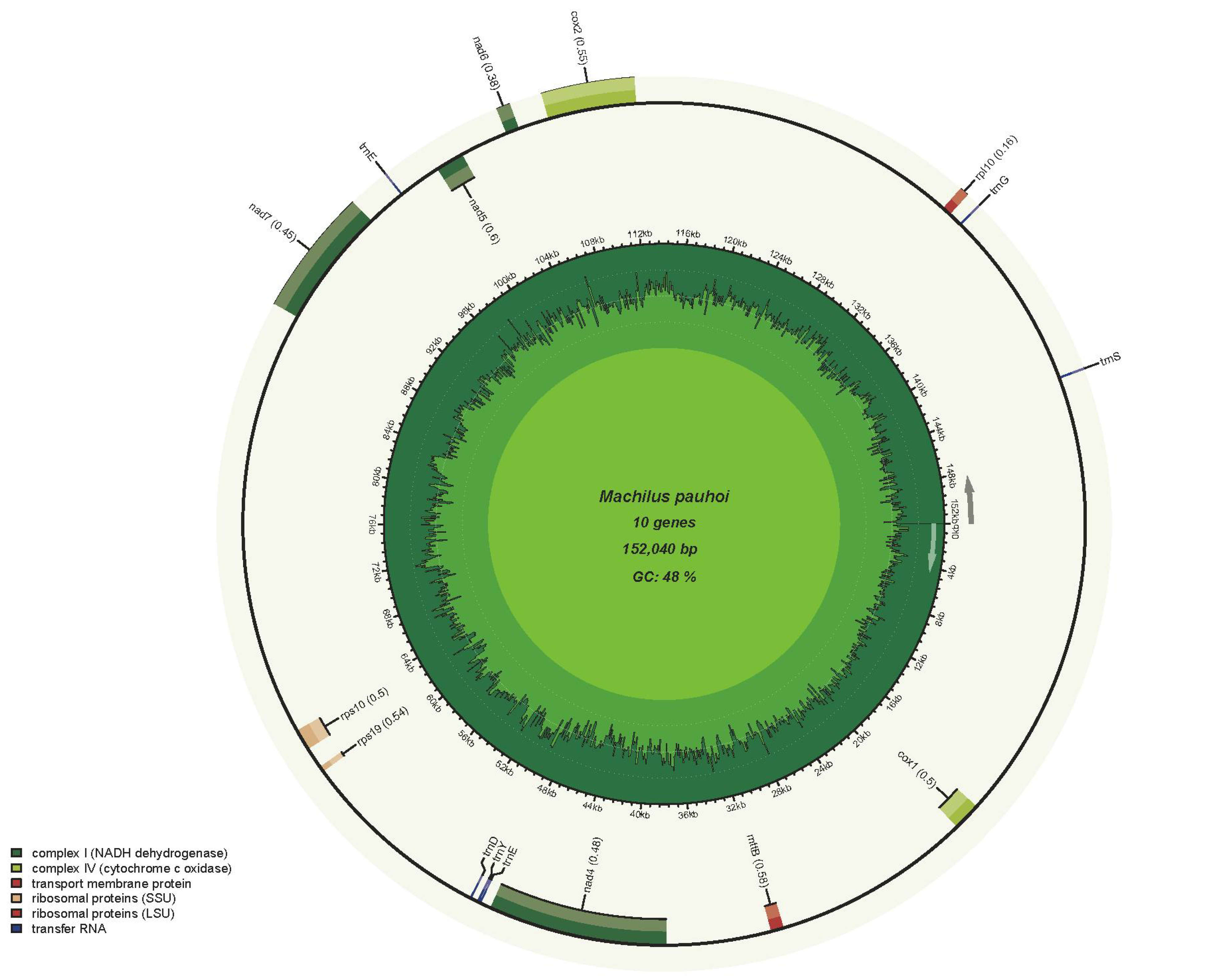
The mitogenome of M. pauhoi was found to have a total length of 775,630 bp, comprising two primary circular structures. The nucleotide composition of the genome exhibited A (26.46%), T (26.45%), C (23.60%), and G (23.49%). The entire mitogenome had a G + C content of 47.09%. The genome structure of M. pauhoi was composed of two circulars with the length of 623,590 bp master member with 33 conserved coding genes and 152,040 bp accessory ring with 10 conserved coding genes (Fig. 1A and 1B), labeled as Chr1 and Chr2, respectively. According to annotation results, the M. pauhoi reference mitogenome contained 68 genes, including 43 protein-coding genes, 22 transfer RNA and 3 ribosome RNA genes (rrn5, rrnL, and rrnS)(Table 1). In addition, we identified 346 open reading frames (ORFs), among which 186 were forward and 160 were reverse encoding OFRs. There were 24 ORFs of length greater than 300 bp. As a member in the ANA grade, the analysis of M. pauhoi (Fig. 2C) mitochondrial genome is helpful to further study of organelle genome in higher plants. We counted and compared the size of the mitochondrial genome of angiosperms (Fig. 2A). Among them, Carex breviculmis had the largest mitogenome, reaching 1,414,795 bp, and Machilus pauboi had only 54.8% of its mitogenome size. The smallest mitochondrial size was Brassica juncea, which was only 219,766 bp. Among the species involved in the comparison, the mitochondrial genome size of M. pauboi was larger than 86% of angiosperms. Hence, significant variations existed in the sizes of mitochondrial genomes across these species. Besides, we also found that there were more circles in the nearby species (Fig. 2B), like Amborella trichopoda (5), Kadsura japonica (8) and Schisandra chinensis (10).
Table 1
Gene composition in the mitogenome of Machilus pauhoi
| group of genes | Name of genes |
| ATP synthase | atp1, atp4, atp6, atp8, atp9(2) |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFC*, ccmFN |
| Ubichinol cytochrome c reductase | cob |
| Cytochrome c oxidase | cox1, cox2**, cox3 |
| Maturases | matR |
| Transport membrane protein | mttB |
| NADH dehydrogenase | nad1*, nad2***, nad3, nad4***, nad4L, nad5**, nad6, nad7****, nad9 |
| Large subunit of ribosome | rpl10, rpl16, rpl2*, rpl5 |
| Small subunit of ribosome | rps1, rps10*, rps11, rps12, rps13, rps14, rps19, rps2, rps3*, rps4, rps7 |
| Succinate dehydrogenase | sdh3, sdh4 |
| Ribosome RNAs | rrn5, rrnL, rrnS |
| Transfer RNAs | trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG, trnI-CAU, trnK-CUU, trnK-UUU, trnL-GAG, trnM-CAU, trnN-GUU, trnP-UGG, trnQ-UUG, trnR-GCG, trnR-UCU, trnS-GCU, trnS-UGA, trnT-UGU, trnW-CCA, trnY-GUA, trnfM-CAU |
| *Intron number; gene (2): number of copies of multi-copy genes |
Repeat sequence analysis in the M. pauhoi and nearby species
A structural model was generated by the long repeat event detection, which were longer than 100 bp. 30 repeat events were found in Chr1 and 3 repeat events in Chr2, respectively. All the repeats in self-circles were under 1000bp. Between Chr1 and Chr2, there were 219 long repeat events. Most of them were under 500bp, and 45 fragments were longer than 100bp. The total length of homologous fragment was 26,474 bp (Fig. 3A), which accounted for 3.4% of the total mtDNA length of M. pauhoi. We found only one long repeat, 8775bp, appearing between two chromosomes (Fig. 3B).
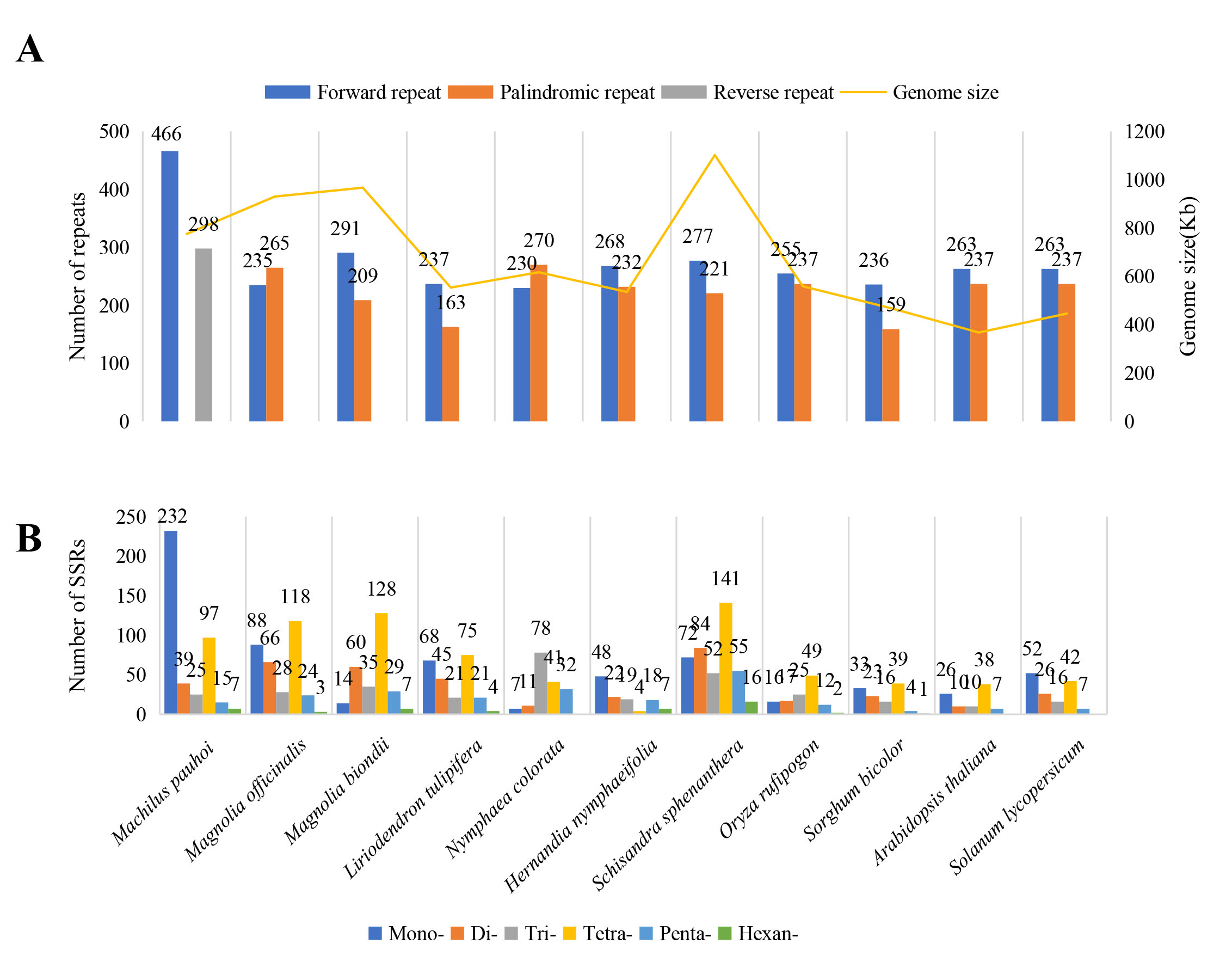
There were great differences in the types of nucleotide repeats among species (Su-Figure 3A). In the M. pauboi mitogenome, a total of 978 dispersed repeats were identified, including 476 forward repeats, 57 reverse repeats, 27 complement repeats and 418 palindromic repeats. The dispersed repeats were mainly forward and palindromic repeats. However, there was no significant correlation between the size of the mitogenome and the number of dispersed repeats in this study. Seven sequenced species related to M. pauboi and two model species were selected to identify and compare the dispersed repeats and Simple Sequence Repeats (SSRs) in the mitogenomes (Fig. 1C). We identified and compared the SSRS in the mitochondrial genomes of 10 angiosperms (Su-Figure 3B). As shown in Fig. 1C, the mitogenome of M. pauboi exhibited a prevalence of mononucleotide and tetranucleotide polymers in its SSRs, which was also observed in the majority of the other analyzed mitogenomes. The number of hexanucleotide repeat was the lowest in these species. Besides, hexanucleotide was not detected in Nymphaea colorata and Arabidopsis thaliana.
Codon preference in this mitogenome and and RNA editing
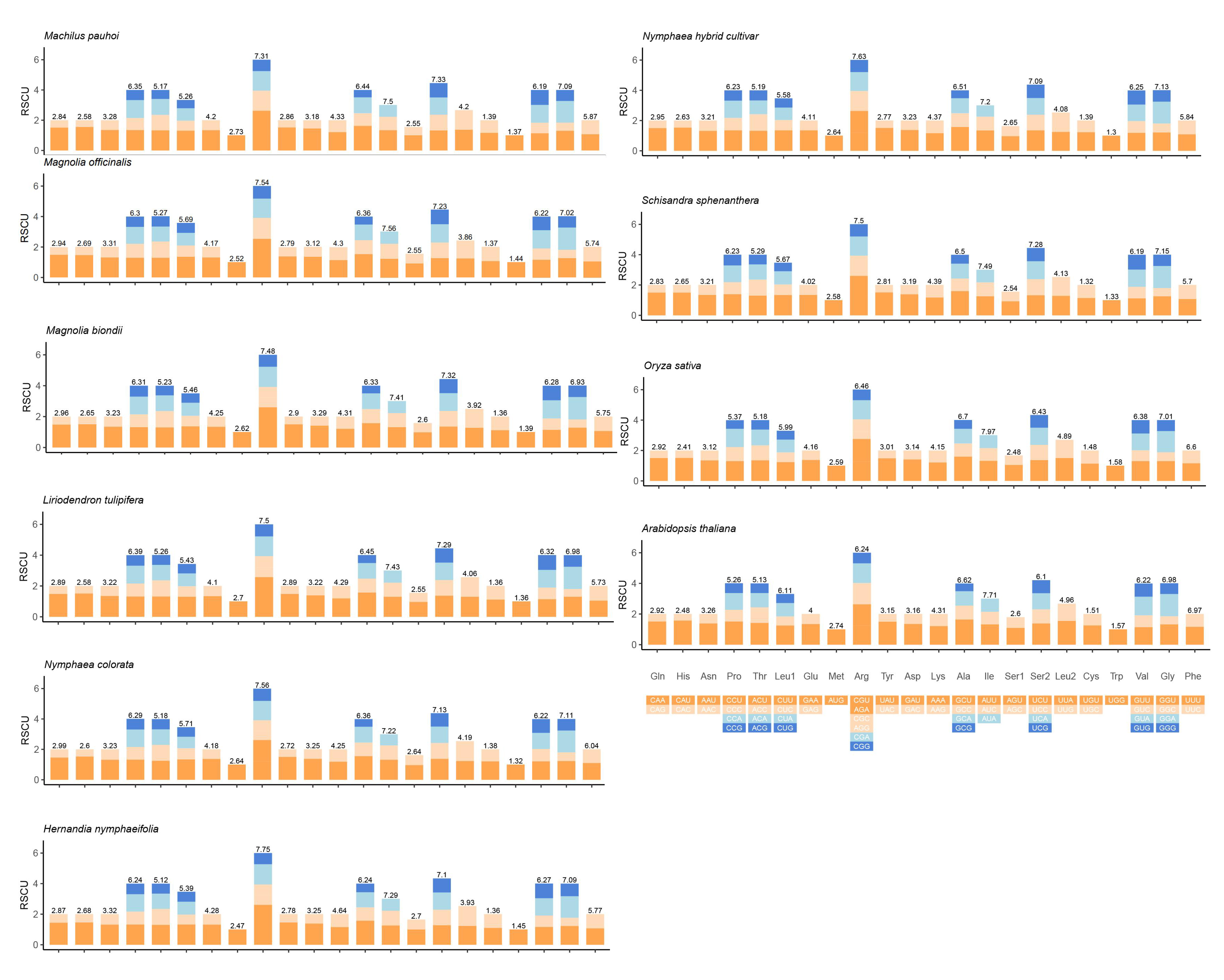
The analysis of synonymous codon usage (RSCU) was conducted on the mt genome of 10 angiosperms, as depicted in Su-Figure 4. In the M. pauhoi mt genome, there were 31 codons with RSCU values greater than 1, indicating a higher frequency of usage compared to other synonymous codons. Among these, a total of 28 codons ended with either A or U bases, accounting for approximately 63.28% of all codons observed. This suggests a slight preference for specific codons within the M. pauhoi mt genome. In all species, the most abundant codon families were Arg and the least was Trp, indicating that the angiosperms mt genome had a similar usage pattern, and RSCU were conserved during the process of evolution.
RNA editing occurs after the transcription process, and the nucleotide deletion, insertion or replacement occurs in the mRNA molecule, which changes the information of gene transcripts. The PREPACT3.0 website predicted the RNA editing sites of PCGs in the mitochondrial genome (Fig. 4). 719 C-to-U RNA editing sites were detected in the PCGs of the mitogenome of Machilus pauhoi (Fig. 4A), which were similar to those of H. nymphaeifolia, L. tulipifera and M. biondii, while 1067 RNA editing sites of M. officinalis were significantly different from those of the other four species. Over 65% of the non-silencing RNA editing sites within the protein-coding genes (PCGs) across the five species were observed at the second position of the codon, while the remaining sites were found at the first position (Fig. 4B). In addition, the level of RNA editing in the first and second position of the same codon was consistent across all five species. We observed a higher number of RNA editing sites in nad4, ccmFn, nad7, cox1, and nad5 (Fig. 4C), while fewer sites were detected in rps1, rps12, rps19, and atp8. The mitochondrial genome of A. planetus exhibited 719 RNA editing sites within its protein-coding genes, resulting in significant alterations to 14 amino acids (Fig. 4C). As shown in the figure, changes in most RNA editing sites lead to the conversion of serine and proline to leucine, causing increased hydrophobicity of the encoded protein. In addition, glutamine and arginine changed less toward the stop codon.
Ka/ Ks analysis among nearby species and Lauraceae
To investigate the evolutionary pattern of PCG in M. pauhoi, the key representative plant "Oryza sativa" of the branch node of Camphor differentiation in the developmental tree was selected as an external reference, and the Ka/Ks value was calculated using 15 PCGs shared by plants in the tree (Fig. 4D,E and F). As shown in the figure, the Ka/Ks ratio of ccmB genes (Hernandia nymphaeifolia, M. pauhoi, Liriodendron tulipifera) in only two species of Camphor order exceeded 1. Most of the other genes had low Ka/Ks ratios (around 0.2–0.8), suggesting that they had undergone purification selection. These findings imply that ccmB genes might demonstrate unique stress resilience when subjected to selective pressure. Similarly, the average value of atp1, cox1 and nad4L Ka/Ks was also the lowest among all genes (about 0.2), and the purification selection of genes was the most obvious. They were very conservative in the evolution process of PCGs in plant mitochondrial genome, which may play an indispensable role in function.
Phylogenetic analysis of M. pauhoi and other species between mitochondria and chloroplast
A mitochondrial phylogenetic analysis was conducted using the conserved protein-coding genes shared by two ANA Grades (N. colorata and N. hybrid cultivar), two monocotyledons and 33 dicotyledons (Fig. 5C). The obtained tree topology was satisfactory and aligned with the APG IV plant classification system(Fig. 5A and B), with a majority of nodes exhibiting self-developing values exceeding 70. The findings indicated that the Laurales consisting of M. pauhoi and H. nymphaeifolia clustered well with the L. tulipifera, M. officinalis, and M. biondii in Magnoliales, which together constitute the Magnoliaceae branch (Fig. 5D). The overall structure of the phylogenetic tree derived from analyzing the complete chloroplast genome was satisfactory and generally aligned with the plant classification system proposed by APG IV, with maximum likelihood (ML) self-developing values of 100 for all nodes. However, the phylogeny of three orders differed from the evolutionary relationships shown by the APG IV plant classification system. The Poales, Asterales, and Myrtales, showed the most pronounced changes in Asterales. The phylogenetic analysis of APG plant classification system is based on nuclear gene fragments, and there are great differences between the fragments, which may be the reason for the inconsistent phylogenetic relationship between them.
Gene rearrangement in M. pauhoi and Lauraceae species
The structure of mitogenome varied greatly among different species, which resulted in the phenomenon of gene rearrangement, widely existing in the mitogenome. We compared the mitogenome gene arrangement patterns of M. pingii with those of four other Magnolia species (H. nymphaeifolia, M. biondii, M. officinalis and L. tulipifera) (Fig. 6A and B). The arrangement pattern of COX-3-ATP8-CYTB fragment was only found in M. pingii and M. biondii, which showed consistency with different patterns in the rest of the species. This indicates that COX-3-ATP8-CYTB fragment was at least inverted and displaced. In addition to the COX3-ATP8-CYTB fragment, the arrangement patterns of other genes were chaotic, which fully illustrated that the mitogenome of Magnolia species underwent gene rearrangements during evolution.
Expression of genes and orfs in mitochondrial genome
There were 41 genes used in the quantification of gene expression. As for M. pauhoi, secondary metabolism manifested active in leaves and the process of wood forming. Thus, we selected four kinds of tissues to examine the conserved gene expression in mitochondrial genome. Most genes obtained a relative low expression compared with TPMs of chloroplast genes. The total expressions of mtgenes displayed a two-type differentiation trend, especially 13 out of 41, part mtgenes still showed high expressions (Fig. 6E). The average TPM of these 13 genes ranged from 161 to 2061. By contrast, the other 28 genes got a ranging TPM from 11 to 330. In the Fig. 6C and D, we analysed the whole TPM distribution of all 41 mtgenes, atp8, rps19, rps10, ccmC, atp9, rps12 and rps1 expressed higher than other genes in both branch tissue and leaves. However, in the no green tissues, these mtgenes got lower expressions than in leaves.
Moreover, we detected the expression of orfs in the mitogenome. Most orfs got regular expression features and relative high TPMs (Fig. 7A), this may infer an organelle genome-specific expression mode and functional regulation (Fig. 7B). Both B18 and B28 families got similar expression pattern, and between cell types, the correlation displayed gathered, in phloem and cambium. On the whole, the expression abundance of the same type of cells was similar in these cells. However, in the expression trend analysis, special expresion orfs can be detected among different cells and different ages of same cells like the results in Fig. 7C. Besides, between different M. pauhoi family, the specific orf got changed (Fig. 7C and D).
Homologous sequence analysis between cytoplasmic genomes and nuclear genomes
By comparing the mitogenome and chloroplast genomes in M. pauhoi, we found 34 homologous sequences (Fig. 8A), totaling 23438 bp, which is 3.02% of the mitochondrial genome. Among them, the homologous sequences of 1-100 bp were the most, with 12, followed by sequences of 301–500 bp, with 7 (Fig. 8B). The longest sequence was 3712 bp (Fig. 8C), located on mitogenome 1, and the shortest sequence was 32 bp, located on mitogenome 2. At the same time, a total of 26 homologous sequences were found on mitogenome 1, with only 8 homologous sequences located on mitogenome 2. Through annotation, we found that many chloroplast annotation genes were transferred to mitochondrial genome, including one protein encoding gene petN and 12 tRNA genes (trnH-GUG, trnR-UCU, trnD-GUC, trnY-GUA, trnE-UUC, trnW-CCA, trnP-UGG, trnI-CAU, trnA-UGC, trnI-GAU, trnN-GUU, trnN-GU_copy2) (Fig. 8C).
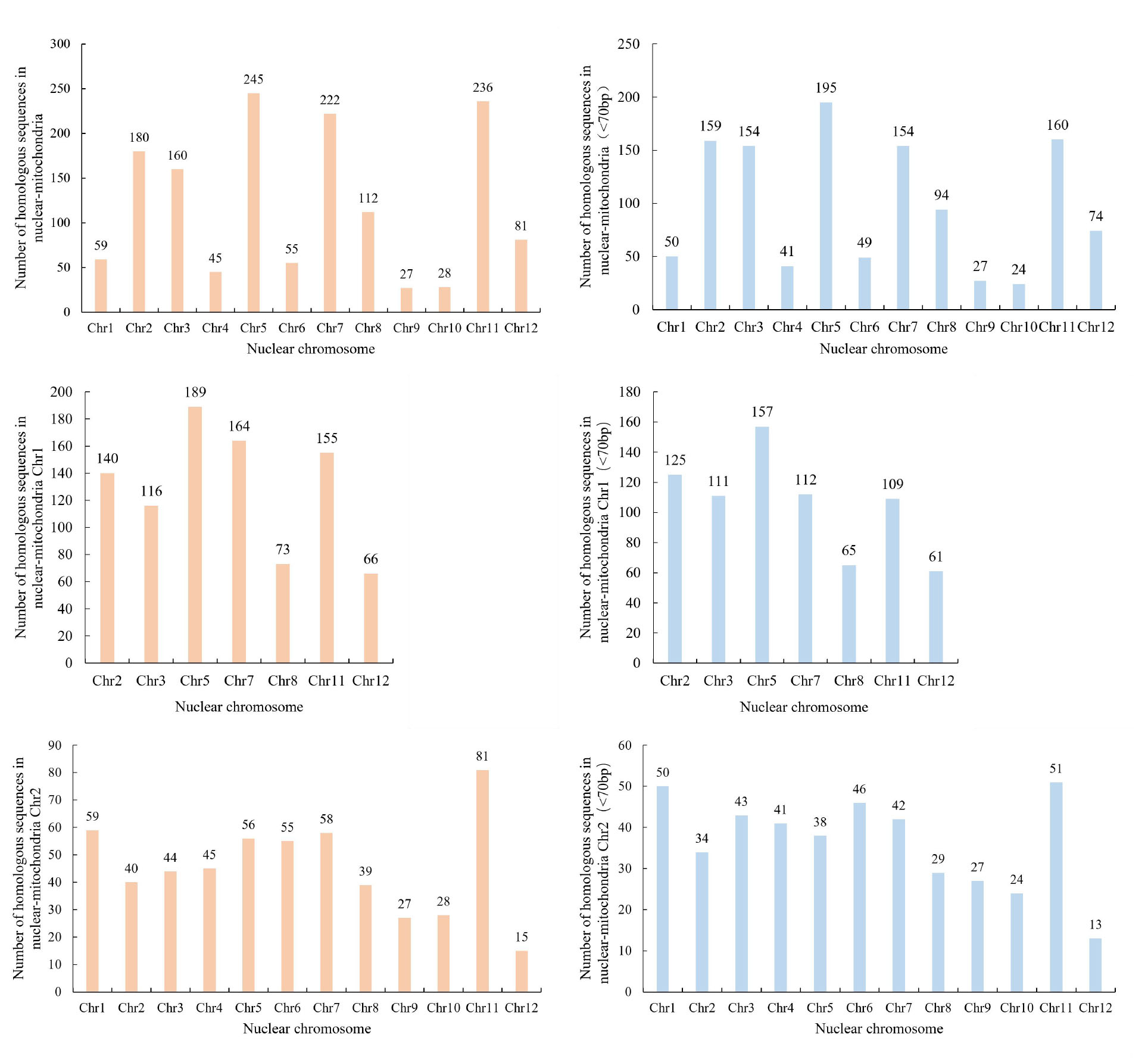
A comparison between the mitochondrial and nuclear genomes of M. pauhoi revealed a discovery of 1450 homologous sequences(Fig. 9A), encompassing a total length of 632,191 bp. (Fig. 9B). Through screening, 1181 homologous sequences with > = 70bp were found, totaling 631964bp. Among the sequences with > = 70bp, the longest homologous sequence was located on mitochondrial Chr 1 at 29292bp, while the shortest sequence was located on mitochondrial Chr 2 at 72bp. The number of sequences with 101-200bp was the highest, with a total of 452, while sequences with 5001-10000bp and > 10000bp had the lowest number, with only 12. It is worth noting that these homologous sequences seem to have chromosomal preference. For example, mitochondrial Chr 2 contained homologous sequences with all chromosomes in the nuclear genome (Chr1-Chr12), while mitochondrial Chr 1 only showed homologous relationships with some chromosomes in the nuclear genome (Chr2, Chr3, Chr5, Chr7, Chr8, Chr11, Chr12) (Fig. 9B). Even though mitochondrial Chr 1 shares homologous sequences with only a portion of the nuclear genome, its number of homologous sequences is still greater than that of mitochondrial Chr 2. At the same time, we also found that the nuclear genome and mitogenomes of M. pauhoi shared or transfered multiple genes through large fragments (Fig. 9C), such as cytb (Mp055539, Mp028253), mRpL2 (Mp026630), mRpS3 (Mp026631), ATP9 (Mp028254), cytc (Mp042170), and mRpS11 (Mp042181).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}