According to previous speculation,age-related hearing loss is primarily attributed to the degeneration of Stira vascularis (SV), which plays a crucial role in maintaining the electrical potential of the inner ear and facilitating the conversion of mechanical signals into electrical signals in the cochlea. (23)。Recent research has identified that cochlear hair cells (HC) are pivotal in determining ARHL. In their autopsy study involving 120 cadaver patients, Liberman et al. observed a significant correlation between hair cell loss and the extent of hearing damage, while stira vascularis were found to be unrelated to hearing damage levels. This groundbreaking study provided novel insights into the key component responsible for ARHL within human cochlea(24).
Cochlear hair cells are highly energy-consuming cells, primarily relying on the oxidative phosphorylation process of mitochondria for their energy source(4, 25).Mitochondria in hair cells maintain homeostasis through division and fusion, thereby ensuring normal mitochondrial function. In hair cells, mitochondria play a crucial role in regulating oxidative metabolism and generating Reactive Oxygen Species (ROS). The aging of hair cells is characterized by the accumulation of oxidative damage caused by ROS buildup, with mitochondria being the primary site of ROS-induced cellular damage (26, 27). Additionally, mitochondria are involved in various other processes such as signaling, cell differentiation, and regulation of the cell cycle and growth (28). During the aging process of cochlear hair cells, disturbances in mitochondrial homeostasis lead to mitochondrial dysfunction (MD), resulting in alterations in intracellular REDOX levels and subsequent cell death(29, 30).
The human mitochondrial genome is a circular, double-stranded, supercoiled molecule with one to several thousand copies per cell consisting of 16,569 bases (31)。Although mitochondrial genes are also protected by proteins, their protective effect is significantly weaker than that of nuclear DNA, rendering them more susceptible to external genotoxic substances (32)。Accumulation of ROS in hair cells also induces mutations in the mitochondrial genome, leading to damage in mitochondrial DNA. Mutation and deletion of mitochondrial mtDNA play a crucial role in hair cell senescence. Markaryan et al. discovered that cochlear tissues from elderly humans exhibited deficiency in 4977-bp mitochondria and and the probability of deficiency would increase with age (33)。Enhanced protection of mitochondrial DNA could be beneficial. Jun Li et al. utilized the DNA methylation inhibitor 5-azacytidine to reduce the methylation level of SOD2, thereby decreasing oxidative stress and copy number variations (CNVs) of mtDNA4834 mutations while inhibiting H2O2-induced apoptosis in hair cells (34)。Subsequently, more studies have confirmed the close association between mutation and deletion of mtDNA with age-related hearing loss (ARHL), suggesting an increased risk for ARHL due to its mutation (35)。
A total of 503 DEGs were identified. KEGG pathway enrichment analysis revealed significant enrichment of DEGs in the oxidative phosphorylation signaling pathway, amino acid anabolic signaling pathway, ribosome signaling pathway, peroxisome signaling pathway, and other pathways associated with mitochondrial function. These findings suggest that alterations in these signaling pathways may contribute to mitochondrial dysfunction and subsequently induce ARHL. Following GO functional enrichment analysis of DEGs, Tables present the top ten entries for each of the three Go ontologies. BP primarily encompasses processes related to mitochondrial transmembrane transport, metabolic processes involving reactive oxygen species, assembly of mitochondrial respiratory chain complexes and genes expression within mitochondria, NADH dehydrogenase complex assembly, precursor metabolite and energy production as well as organic acid decomposition and small molecule metabolism. CC involves the formation of components such as mitochondrial membranes, mitochondrial matrixes, myelin sheaths, extracellular exosomes as well as complete components found in other organelle membranes including extracellular vesicles and organelles. MM molecules are involved in activities such as binding to 2- and 4-ferric sulfur clusters or metal clusters; mRNA/tRNA methyltransferase activity; catalytic activity; carboxylic ester hydrolase activity.
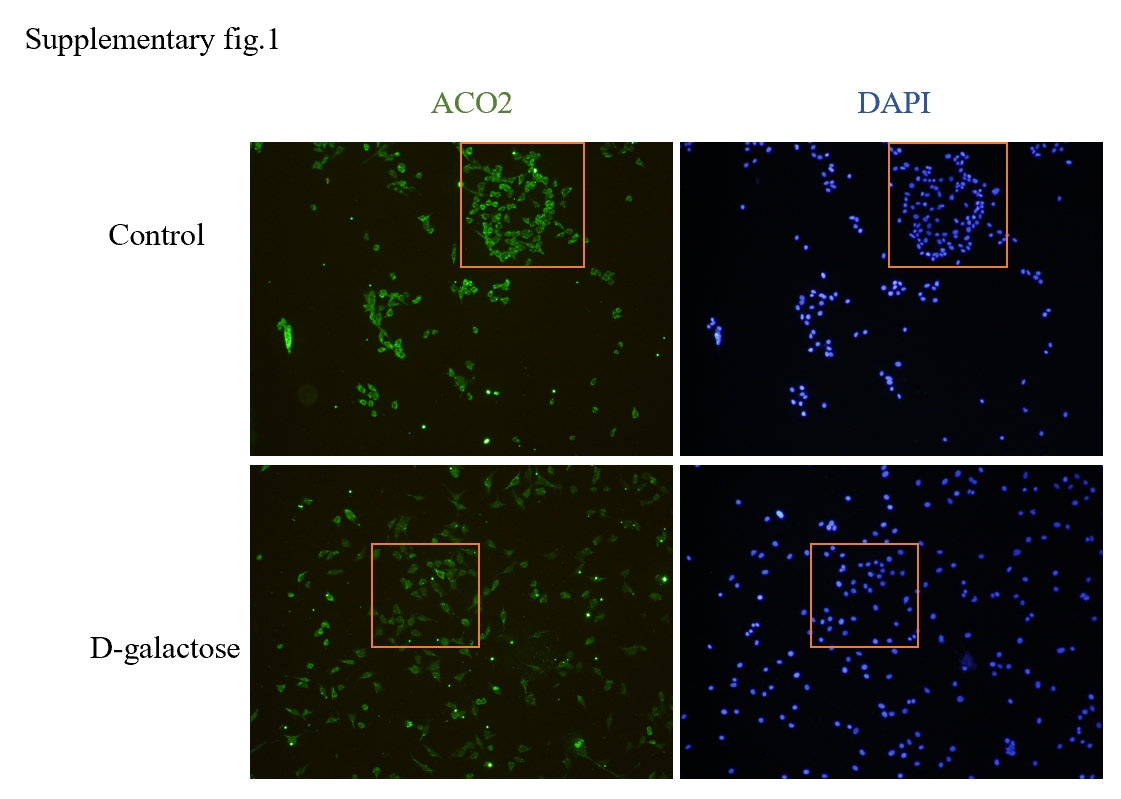
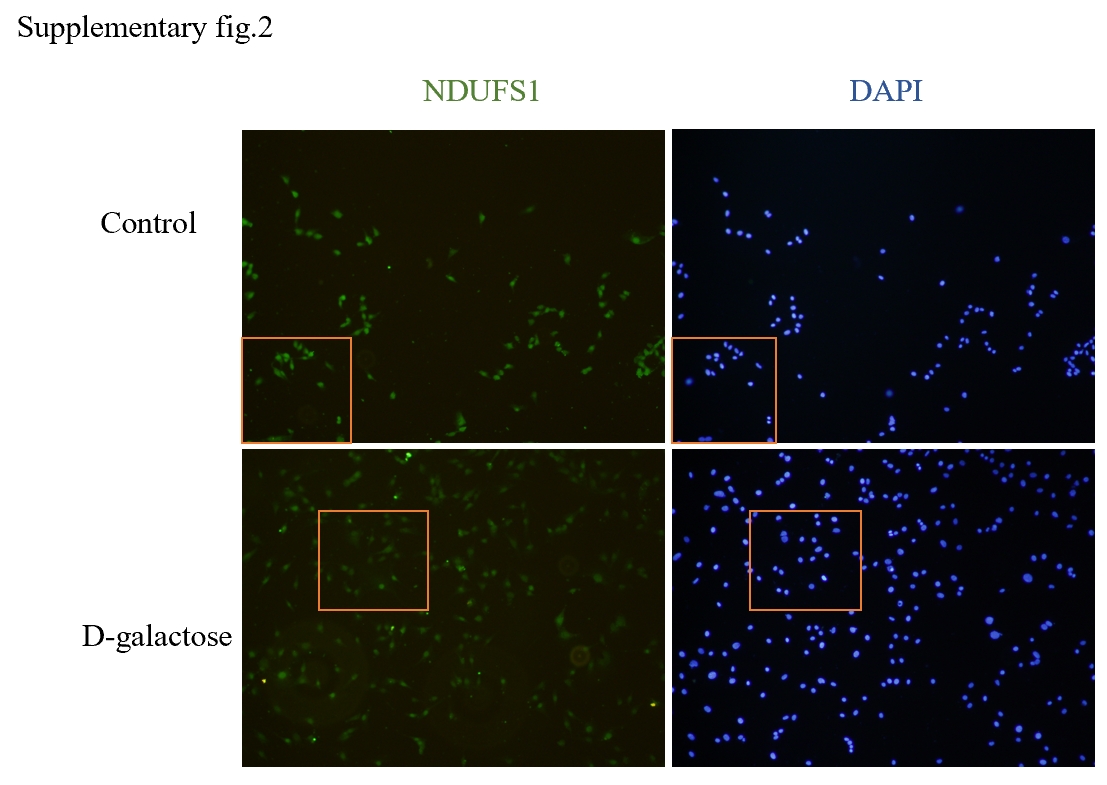
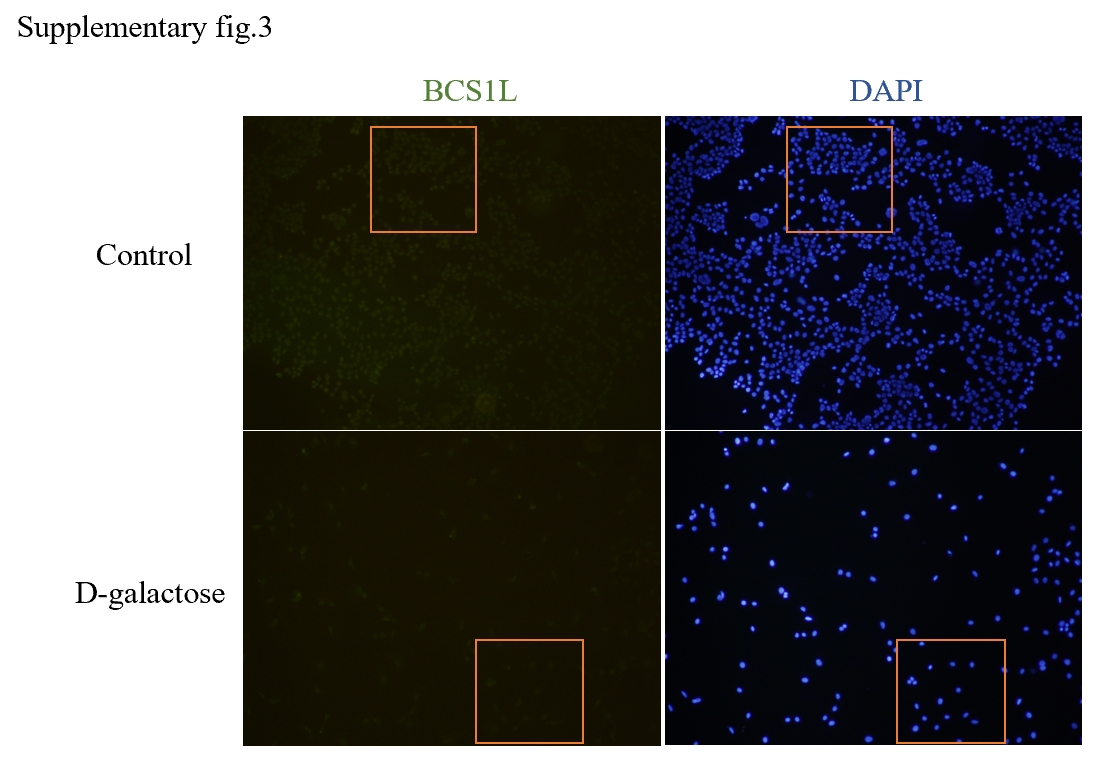
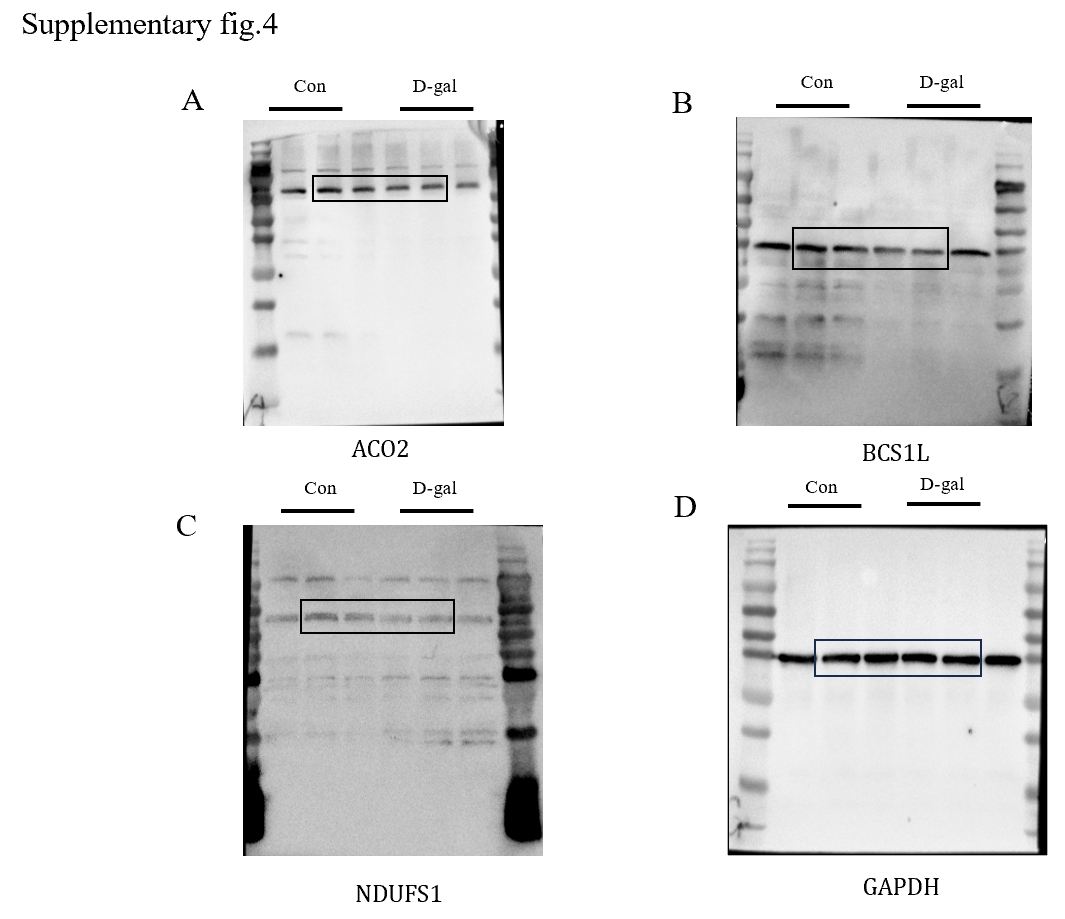
In light of the strong correlation observed between these differential genes and mitochondrial function, we performed an intersection analysis with mitochondrial DNA to identify differentially expressed mitochondrial DNA associated with age-related hearing loss. Among them, Aco2, Bcs1l, and Ndufs1 emerged as three key hub genes closely linked to mitochondrial function.
Mitochondrial aconitase 2 (ACO2) belongs to the iron-sulfur cluster hydratase family. Upon binding to the enzyme structure, the active iron-sulfur cluster catalyzes citric acid reversibly into isocitrate in the TCA cycle and facilitates dehydration and rehydration reactions (36). Aco2 is an indispensable enzyme in the tricarboxylic acid cycle, playing a crucial role in coordinating mitochondrial and autophagy functions for energy metabolism within cellular mitochondrial respiratory chain. ACO2 significantly contributes to cellular energy metabolism, maintenance of iron homeostasis, resistance against oxidative stress, as well as preservation of mitochondrial DNA (mtDNA) integrity (37). Research has indicated that mutations in the ACO2 gene may be associated with premature aging (38). Furthermore, studies have demonstrated that ACO2 impacts neuronal function and survival during aging process, potentially contributing to Alzheimer's disease onset (39).Targeting ACO2 for enhancing energy metabolism could serve as a promising therapeutic strategy for Parkinson's disease and other neurodegenerative disorders(40)。Currently, there are extensive reports on the involvement of ACO2 mutations in energy metabolism issues and progression of neurodegenerative diseases such as optic atrophy, microcephaly, intellectual impairment cognitive decline hypotonia spastic paraplegia(41–43).
As a chaperone and translocation enzyme in the inner mitochondrial membrane, the BCS1L protein plays a crucial role in promoting the final folding and assembly of respiratory complex III by facilitating the insertion of the Rieske iron-sulfur subunit into the complex(44). The functional structure of BCS1L consists of three distinct domains: (a) N-terminal domain comprising three special parts - transmembrane domain (TMD), mitochondrial targeting sequence (MTS), and input auxiliary sequence (IAS); (b) BSC1L-specific domain; and (c) C-terminal AAA-ATPase domain. The interactions between TMD/MTS and BCS1L assist in anchoring proteins within the mitochondrial matrix for subsequent transport (45, 46)。Mutations in BCS1L are commonly associated with defects in human mitochondrial complex III (47), where mutated BCS1L protein disrupts complex III assembly, reduces mitochondrial electron transport chain activity, impairs ATP synthesis, and increases reactive oxygen species production(48), thereby causing damage to cellular components that accelerates aging. In addition to various diseases such as sensorineural hearing loss (Bjornstad syndrome) and severe multisystem organ failure (Complex III deficiency and GRACILE syndrome), mutations in BCS1L also lead to features related to central nervous system dysfunction including movement disorders, seizures, and clinical manifestations resembling Leigh's encephalopathy(49). Furthermore, BCS1L is involved in regulating mitochondrial autophagy; its dysfunction affects damaged mitochondria clearance leading to dysfunctional organelle accumulation which can contribute to cell senescence (44).
NADH:ubiquinone oxidoreductase core subunit S1 (NDUFS1) is the largest subunit within mitochondrial complex I, responsible for catalyzing the initial step of respiratory nicotinamide adenine dinucleotide (NADH) oxidation in mitochondria. It plays a pivotal role in maintaining the stability and functionality of mitochondrial complex I (50).Deletion or mutation of NDUFS1 results in reduced levels and catalytic activity of mitochondrial complex I, disrupting NADH homeostasis and impeding electron transfer within the respiratory chain. Consequently, this leads to substantial intracellular reactive oxygen species (ROS) production(51),impaired mitochondrial function, and ultimately accelerates cellular aging processes. Furthermore, deficiency in NDUFS1 causes loss of mitochondrial complex I, contributing to Leigh encephalopathy (52). Additionally, studies have suggested that mutations in Ndufs1 may be associated with Parkinson's disease and can serve as diagnostic markers for its detection(53). A Danish cohort study has also demonstrated that NDUFS1 reflects physical conditions among elderly individuals by correlating with grip strength and walking speed(54).




{kind=link}
{kind=link}
{kind=link}
{kind=link}