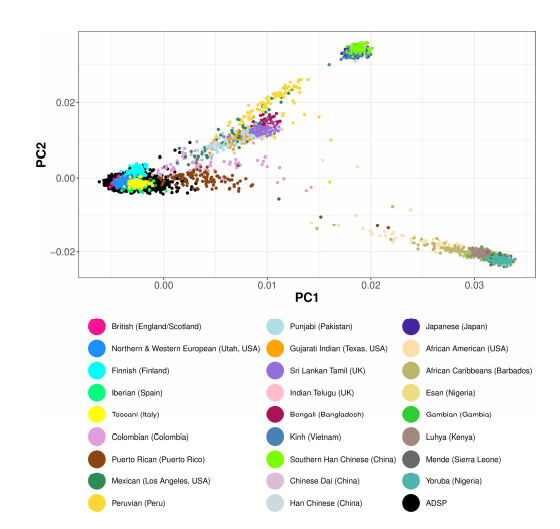
Background: Genetic studies have shifted to sequencing-based rare variants discovery after decades of success in identifying common disease variants by Genome-Wide Association Studies using Single Nucleotide Polymorphism chips. Sequencing-based studies require large sample sizes for statistical power but often inadvertently introduce batch effects because samples are typically collected, processed, and sequenced at multiple centers. Conventionally, batch effects are first detected and visualized using Principal Components Analysis and then controlled by including batch covariates in the disease association models. For sequencing-based genetic studies, because all variants included in the association analyses have passed quality control measures, this conventional approach treats every variant as equal and ignores the substantial differences still remaining in variant qualities and characteristics such as genotype quality scores, alternative allele fractions (fraction of reads supporting alternative allele at a variant position) and sequencing depths.
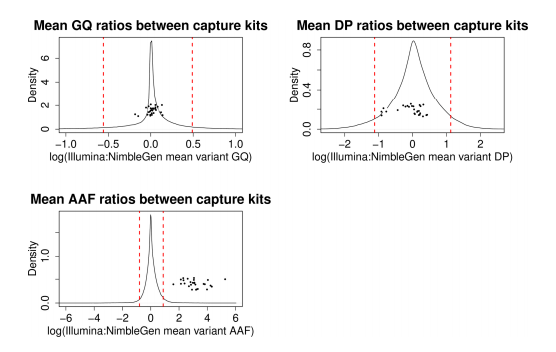
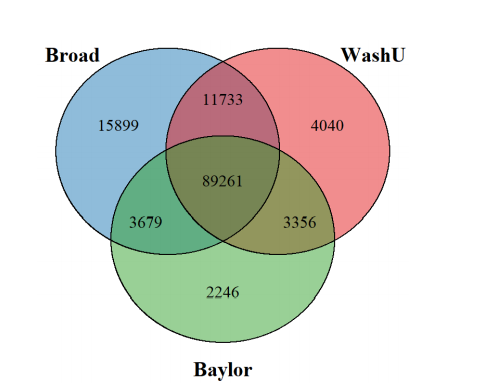
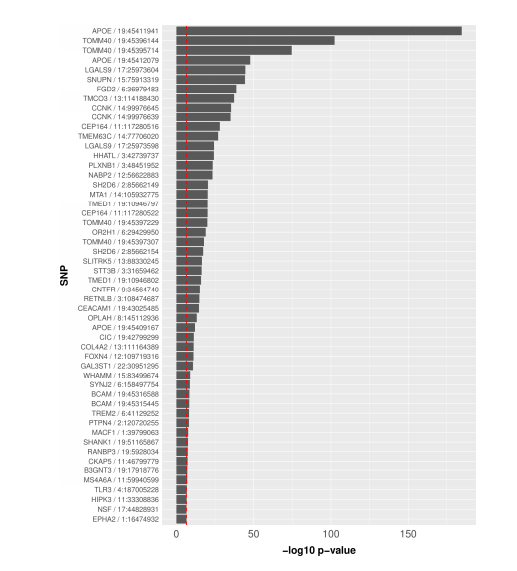
Results: In the Alzheimer’s Disease Sequencing Project (ADSP) exome dataset of 9,904 cases and controls, we discovered hidden variant-level differences between sample batches of three sequencing centers and two exome capture kits. Although sequencing centers were included as a covariate in our association models, we observed differences at the variant level in genotype quality and alternative allele fraction between samples processed by different exome capture kits that significantly impacted both the confidence of variant detection and the identification of disease-associated variants. Furthermore, we found that the association signals of a subset of top disease risk variants came exclusively from samples processed by one exome capture kit that was more effective at capturing the alternative alleles compared to the other kit.
Conclusions: Our findings highlight the importance of additional variant-level quality control for large sequencing-based genetic studies. More importantly, we demonstrate that automatically filtering out variants with batch differences may lead to false negatives if the batch discordance came largely from quality differences and if the variants from one batch had better quality scores.
{kind=link}
{kind=link}
{kind=link}
{kind=link}