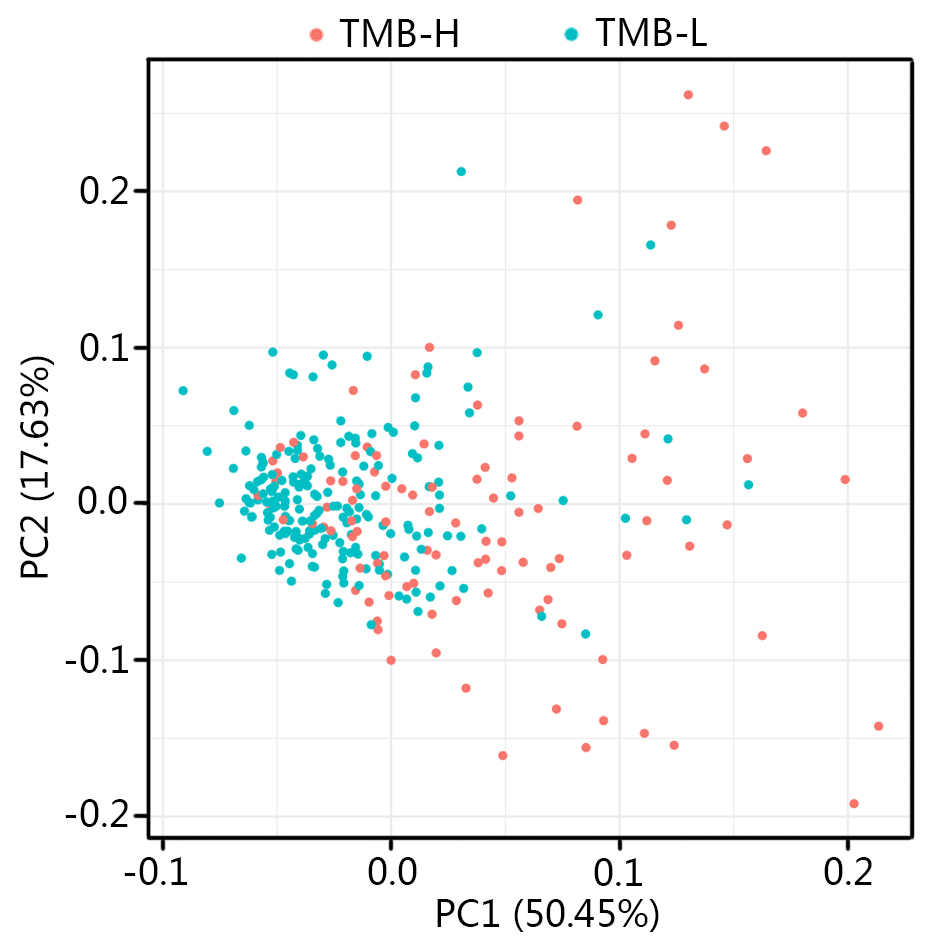
Immunotherapy has been demonstrated particularly successful in NSCLC treatment and is being adopted as a first-line treatment option worldwide [13, 22, 42]. Nevertheless, only a small portion of the unselected patients can benefit from immunotherapy [25, 43]. Therefore, biomarkers for patient selection become important to achieve effective therapy. TMB has been recognized as the effective prognostic biomarker in NSCLC patients according to the results from numerous clinical trials [22, 23, 44, 45]. Although the targeted NGS has been proved to be an alternative approach of WES for the prediction of TMB, the accuracy and simplicity of targeted NGS remain challenging as various parameters should be taken into consideration [46]. In this study, we developed a mathematic multi-omics model that could precisely predict the TMB in patients with LUAD through LASSO logistic regression analysis, and the prediction accuracy of the model was validated in an independent cohort with high sensitivity and specificity (Fig. 8). Furthermore, as the input parameter in this model includes expression profiles of 7 genes, 7 miRNAs, and the methylation profiles of 6 CpG sites, which could be obtained through quantitative real time-polymerase chain reaction (qRT-PCR). This model paved the way for the further development of the simplified qRT-PCR based clinical assay for TMB prediction.
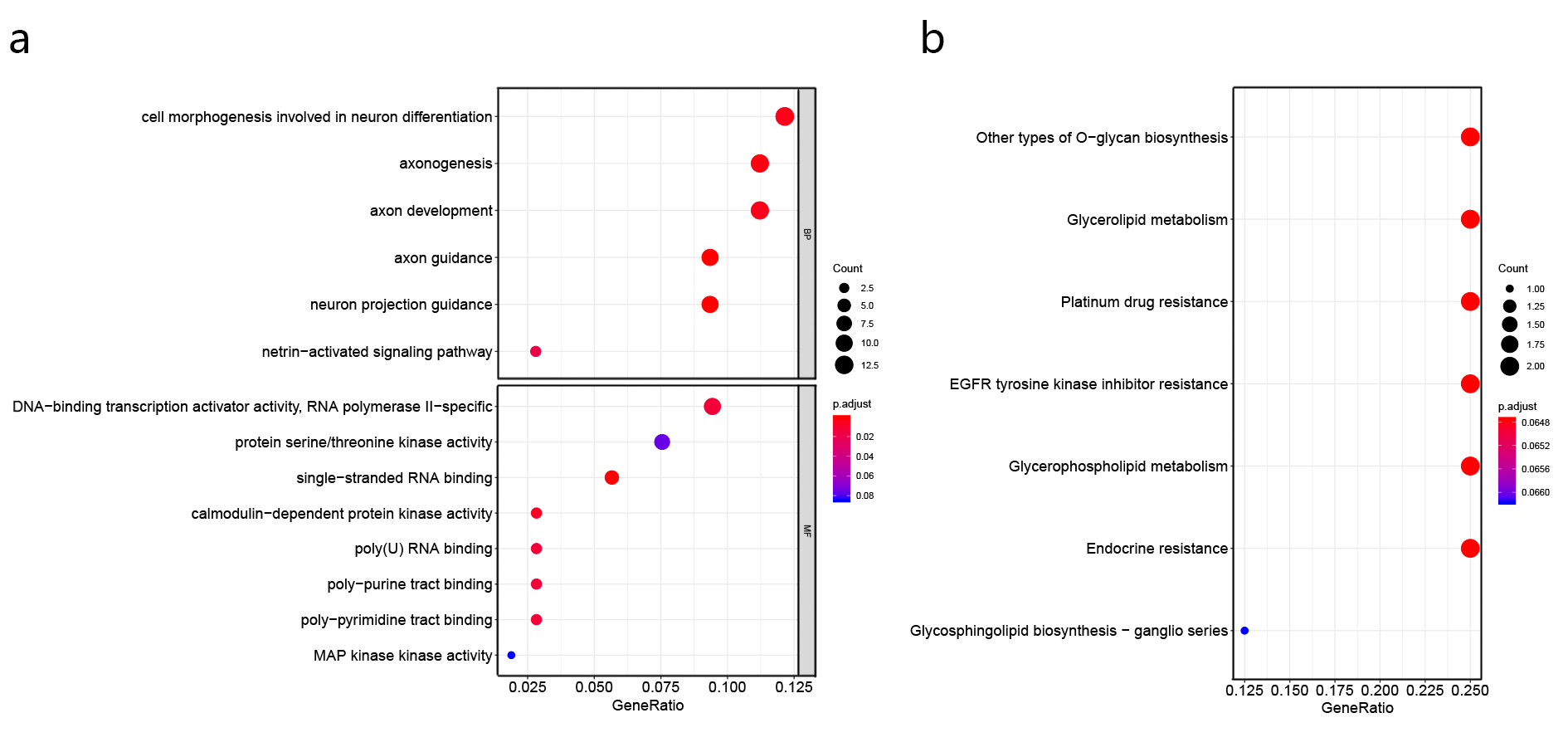
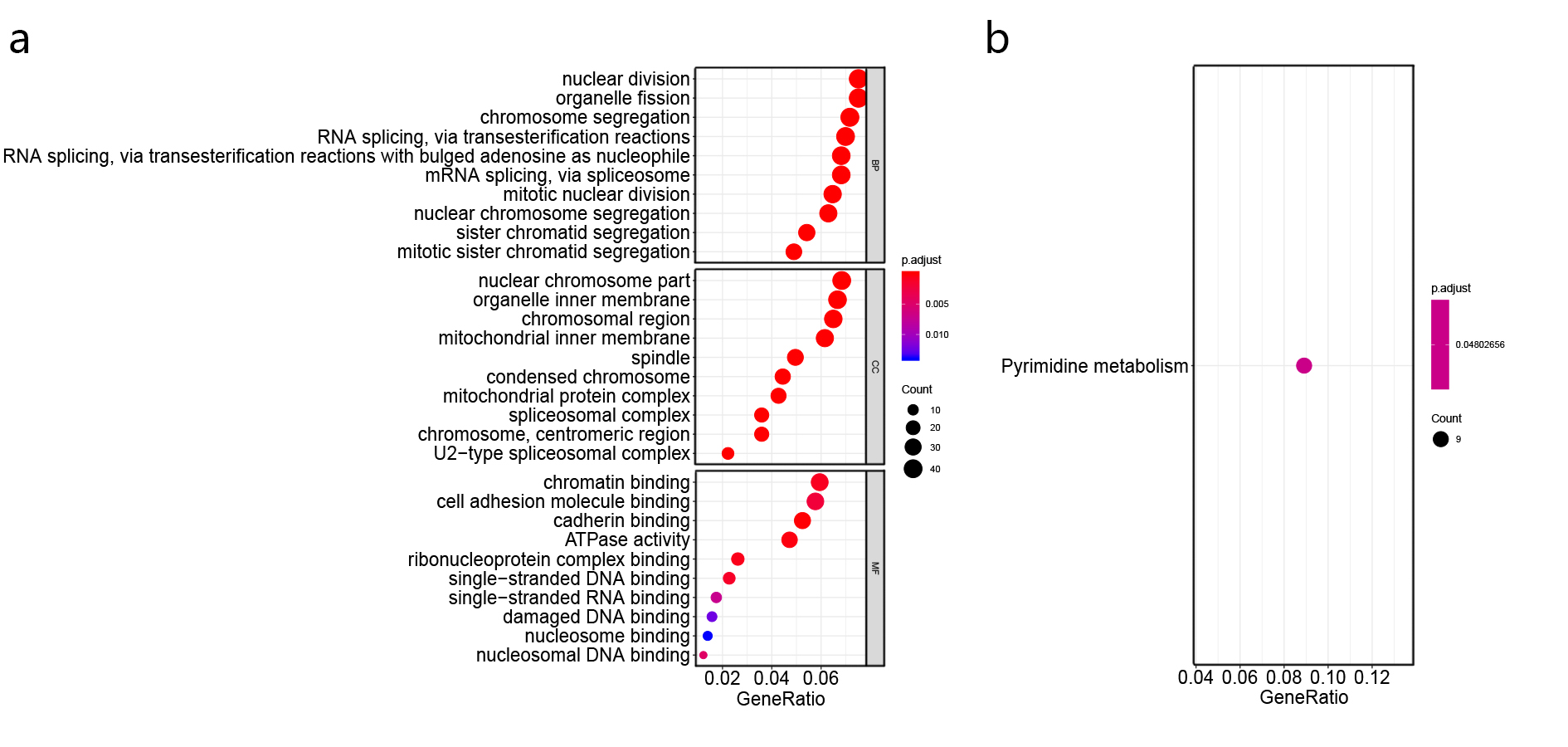
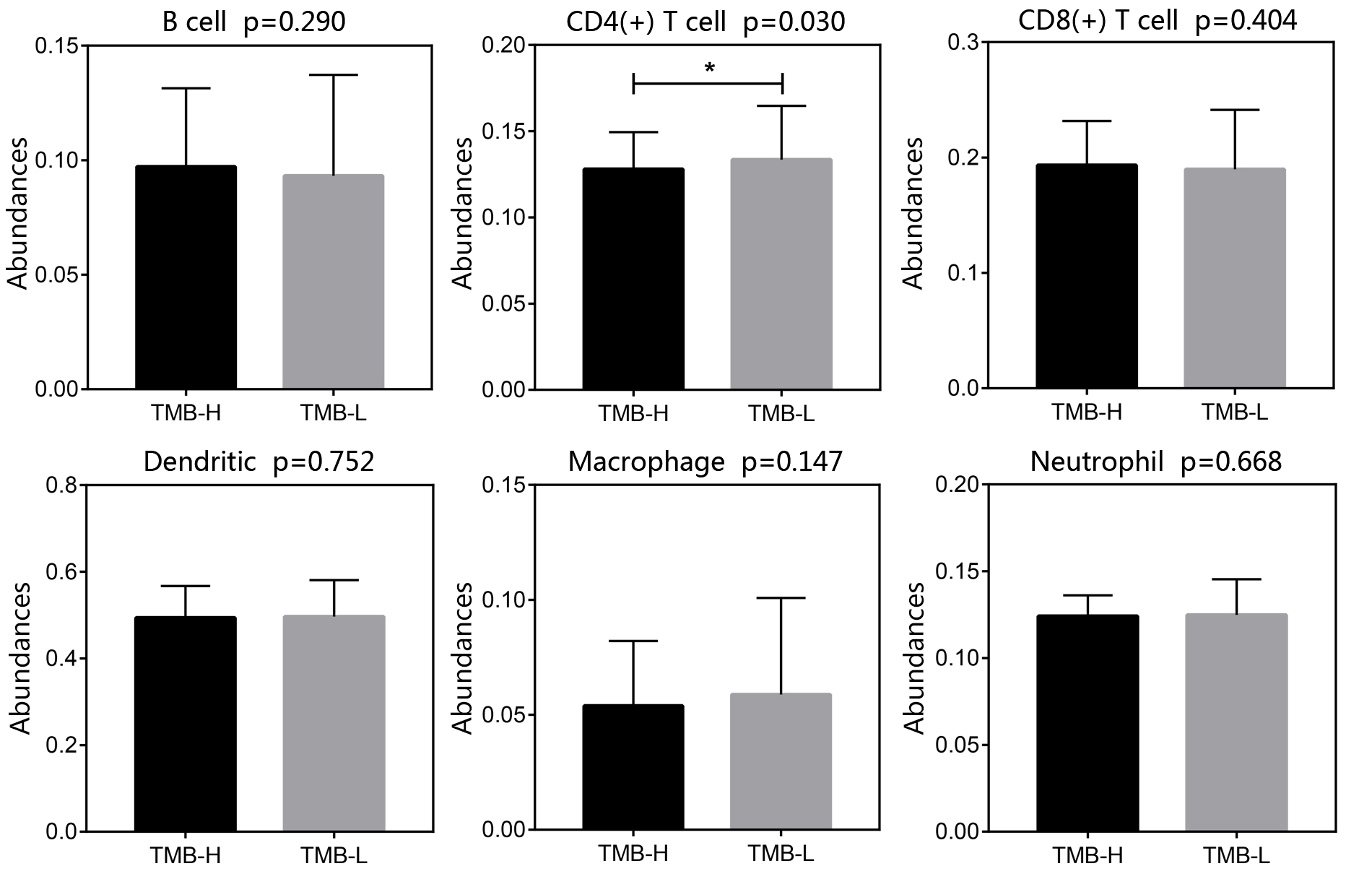
The tumor microenvironment refers to the network of cells and structures that surround a tumor cell, and it consists of immune cells, mesenchymal cells, endothelial cells, extracellular matrix (ECM) molecules, and inflammatory mediators [47]. High TMB indicates the presence of more neoantigens in the tumor microenvironment, which promote the inflammatory response and result in the alteration of transcriptomic and epigenetic signature [47]. It has been proved that gene expression signatures in the tumor microenvironment were associated with the prognosis in NSCLC [48–51]. In agreement with previous studies, the differentially expressed genes between TMB-H and TMB-L patients identified in this study were found to enrich in the immune-related damaged DNA binding, nuclear division, nuclear chromosome segregation, organelle fission, single-stranded DNA binding, ribonucleoprotein complex binding and pyrimidine metabolism (Additional file 2: Figure S2) [52–56]. The 7 genes used in constructing the TPM might be involved in the carcinogenesis, for instance, Y box binding protein 2 (YBX2) was differentially expressed between different subtypes of breast cancer and was one of RNA processing factors which contribute to subtype-specific splicing [57]. Meanwhile, it was found that LINC00958 promoted cell proliferation and migration in oral squamous cell carcinoma through miR-627-5p/YBX2 axis [58]. Helicase‑like transcription factor (HLTF) was a tumor suppressor, the expression of HLTF was negatively correlated with colorectal cancer and its overexpression regulated the TGF‑β/SMAD pathway to suppress the migration and invasion of CRC [59]. It was reported that the wild type alleles of kinesin light chain 3 (KLC3) Lys751Gln was significantly correlated with greater smoking intensity, and genetic variations may influence the progression of lung cancer [60]. Then, WRN helicase interacting protein 1 (WRNIP1) was inhibited by miR-22 to increase the radiosensitivity of small-cell lung cancer cells [61]. Besides, the expression of CDC28 protein kinase regulatory subunit 1 (CKS1B) in lung cancer cells developed the chemoresistance through Hsp90 and MEK1/2 pathway [62].
miRNAs expression in the tumor microenvironment plays a crucial role in mediating and controlling several immune and cell interactions, and convolute in the regulation of immune checkpoints, PD1 and PD-L1 [63]. It was reported that a 25 miRNA-based signature classifier could predict the TMB level with high accuracy [64]. A cluster of highly expressed miRNA including hsa-miR-492, hsa-miR-498, hsa-miR-320 were found to be correlated with tumorigenesis of retinoblastoma [65]. Moreover, the invasion, proliferation and migration of cervical cancer cells were found to be promoted by hsa-miR-6727-5p, which might play an important role in cervical cancer progress [66]. In this study, we mapped the differentially expressed miRNAs between TMB-H and TMB-L patients to their target genes, and enrichment analysis of the target genes suggested that DNA-binding transcription activator, single-stranded RNA binding, MAP kinase activity, Glycerolipid metabolism, Platinum drug resistance and EGFR tyrosine kinase inhibitor resistance related to lung cancer metabolism were affected in the tumor microenvironment. The 7 miRNAs used in constructing the TPM in this study include hsa-miR-571, hsa-miR-586, hsa-miR-151b, hsa-miR-378i, hsa-miR-6727-5p, hsa-miR-502-3p, hsa-miR-6798-3p. It was reported that hsa-miR-378i, hsa-miR-92a-3p, hsa-miR-93-5p and so on were important for identifying both colon and rectal cancer [67]. In previous study, it was demonstrated that gallbladder carcinoma metastasis was developed by lncRNA-HGBC through the activation of miR-502-3p-SET-AKT cascade [68].
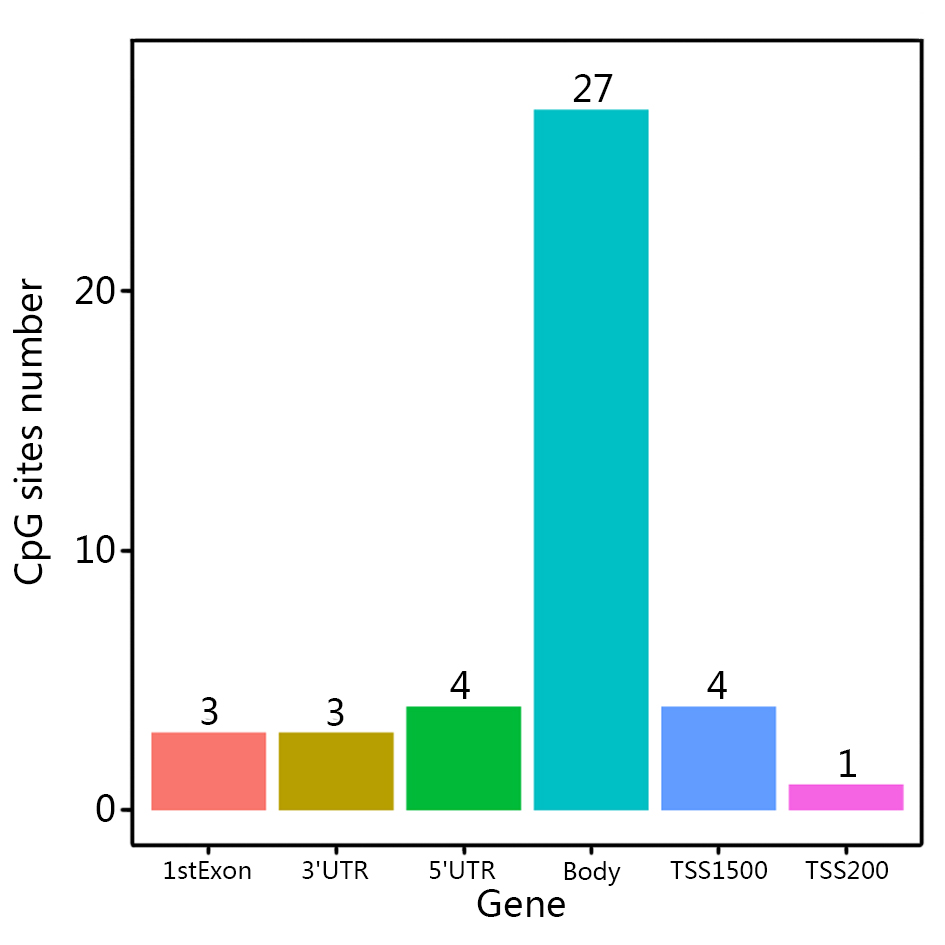
Changes in DNA methylation is one of the most important epigenetic alteration in tumor microenvironment. A multicenter study in 15 hospitals suggested epigenomic profile based on a microarray DNA methylation signature (EPIMMUNE) could serve as an effective biomarker in predicting the outcomes of NSCLC patients treated with PD-1 inhibitors [69], and the FOXP1 could be associated with validated predictive biomarkers for better-selecting patients to benefit with immunotherapy [69]. The CpG sites signature also had a relatively high predictive performance (measure = 0.861) of TMB, suggesting its great value in NSCLC prognosis. Cg02849937 located in the TSS1500 region of C7orf13, and its expression level was inverse associated with promoter methylation using whole-genome integrative analysis [70]. In addition, cg27281030 located in the TSS1500 region of NLRP12. Previous studies demonstrated that NLRP12 could regulate inflammation and tumor, and it is believed that hepatocellular carcinoma was negatively regulated by NLRP12 through the suppression of inflammation and proliferation of hepatocytes [71]. Besides, cg23179456 located in the TSS1500 region of ADCY4, which has been proved to be a biomarker for breast cancer [72].
Through multi-omics analysis, we integrated gene or miRNA expression and DNA methylation data to reflect the subtle alterations of the tumor microenvironment to precisely predict the TMB for better prognosis of patients with LUAD in immunotherapy. Fragments per kilobase per million mapped reads (FPKM) of YBX2, HLTF, KLC3, WRNIP1, CKS1B, RNF26, ZYG11A, RPM of hsa-miR-571, hsa-miR-586, hsa-miR-151b, hsa-miR-378i, hsa-miR-6727-5p, hsa-miR-502-3p, hsa-miR-6798-3p as well as beta-value of cg02031308, cg03286742, cg04046889, cg12095807, cg16794961, cg24553235 were extracted from the RNA-seq, miRNA-seq and Illumina HumanMethylation450 BeadChip respectively for calculating the TPM score, and the threshold of TPM for predicting the TMB level had been predetermined as -2.947 with the specificity of 0.887 and the sensitivity of 0.825 in the training cohort. Although the FPKM, RPM and beta value involved in the TPM were based on high-throughput sequencing or chip analysis, it is feasible to convert them to cycle threshold (Ct) value in qRT-PCR, and thus simplify the prediction of TMB by using benchtop qRT-PCR instrument. The conversion of FPKM in different samples to Ct values could be probably through the comparison of the targeted gene expression to reference gene expressions, such as actin and eukaryotic elongation factor (eEF), which have relative consistent expression under different tumor microenvironment, and the beta value of CpG sites could also be converted into Ct value though the quantitative MethyLight technology [73]. To our best knowledge, this is the first time to construct the TPM for patients with LUAD from multi-omics view.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}