SIRT6 Depletion Results in a Shift in Tryptophan Metabolism
We took advantage of murine embryonic stem (ES) cells metabolomics data40 of WT vs. SIRT6 KO mice and analyzed it to test if SIRT6 dependent changes associate with metabolite sets reported in CSF from various diseases as proxy for brain health.
The analysis revealed enrichment associated with degenerative, psychiatric, and brain infection disease (Figure 1A). Interestingly, among the associated metabolites for AD, dementia, schizophrenia and brain inflammatory diseases, are tryptophan and its derivatives- kynurenic acid and quinolinic acid. This overlap was interesting, since tryptophan and its derivative metabolites are known biomarkers in the CSF for degenerative and psychiatric diseasese7,44–48 and SIRT6 deficiency could recapitulate in a significant manner these metabolic signatures. Next, we analyzed ES cells metabolomics data40, focusing on tryptophan and its catabolites. Our results indicated significant changes in various metabolites, including an increase in tryptophan and its derivatives levels such as kynurenic acid and quinolinate in SIRT6 KO cells. (Figure 1B-D). These results were validated in two additional human cell lines, neuroblastoma-derived SHSY-5Y and adenocarcinoma-derived HeLa SIRT6 WT and KO cells. In all experimental models examined, the absence of SIRT6 resulted in an augmentation of tryptophan levels (Figure 1D, Suppl. Figure 1A). Since Tryptophan is an essential amino acid we tested the levels of its transporter Slc75a, (the most abundant tryptophan transporter in the brain49,50) and found an increased expression in the brains of brS6KO mice (Figure 1E).
Furthermore, directed metabolomic analysis of the SIRT6 KO in SHSY-5Y, HeLa cell lines showed increase in anthranilic acid along with a reduction in serotonin levels (Figure 1G, Suppl. Figure 1B), suggesting a shift favoring the kynurenine pathway (Figure 1H). However, the levels of NAD+, measured in ES, were also reduced, even though its precursor quinolinate was upregulated (similar to the phenomena observed in aging51,52, Figure 1C).
We performed untargeted metabolomic analysis of SIRT6 deficiency in SHSY-5Y (representing a similar to neuron line), HeLa (representing cancer line), and ARPE19 (normal retina-pigmented epithelium53) cell lines, using principal component analysis. The metabolome differed between cells that did and did not express SIRT6, but the scope of differences depended on the cell type (Suppl. Figure 1C-G). Importantly, in HeLa cells, tryptophan metabolic pathway was one of top 10 categories contributing to differences between control and SIRT6 KO cells (Suppl. Figure 1D).
Overall, SIRT6 affected tryptophan levels and its catabolic byproducts in mouse and human cell lines, suggesting a conserved basic mechanism across animal and cell types that shift the metabolites toward the kynurenine pathway (Figure 1H).
SIRT6 Regulates Transcription of Genes Involved in Tryptophan Catabolism
To investigate whether these changes are associated with gene regulation, we conducted a transcriptomic analysis of brains from full-body SIRT6 knockout (SIRT6 KO) and brS6KO mice, using microarray and RNA-seq methods, respectively40,41. This analysis revealed significant differences between SIRT6 KO and wild-type mice in the expression of genes involved in tryptophan metabolic pathways (Figure 2A-B). The hypergeometric test indicates correlation in the results of the two separated models emphasizes the similarity between the datasets show how fundamental SIRT6 role in tryptophan metabolism is (Figure 2B). In addition, we performed a quantitative PCR with additional animals and using probes for genes that were not detected in the transcriptomic experiments. Importantly tryptophan 2,3-dioxygenase 2 (TDO2; predominantly expressed in the brain54) and indoleamine 2,3-dioxygenase (IDO1 and IDO2; predominantly expressed in immune cells55,56), both encoding rate-limiting enzymes involved in the conversion of tryptophan to kynurenin57,58 were upregulated. Additionally, the levels of kynureninase (Kynu) mRNA were significantly elevated in the brains of brS6KO mice versus wild-type controls, whereas the expression of kynurenine 3-monooxygenase (Kmo), which is crucial for energy production, was diminished (Figure 2C). No significant changes were observed in the expression levels of Ccbl1, Haao and Ddc (Figure 2C).
In the serotonin-melatonin pathway, tryptophan hydroxylase 1 (Tph1) expression, which catalyzes the initial step in melatonin production, was upregulated in the brains of brS6KO mice versus controls. In contrast, the expression of rate-limiting enzymes aralkylamine N-acetyltransferase (AANAT) and N-acetylserotonin O-methyltransferase (Asmt) were downregulated (Figure 2C-D).
Using ChIP-seq and ChIP-qPCR, we confirmed that SIRT6 binds directly to the promoter regions of TDO2 IDO1, and AANAT (Suppl. Figures 2A-B). These results suggest that SIRT6 balances the expression levels of the rate-limiting enzymes of the two pathways, and in the absence of SIRT6, tryptophan would be directed to the Kynurenine pathway, at the expense of serotonin/melatonin pathway.
Serotonin and Melatonin Production and their corresponding pathways are affected in brS6KO Mice
Changes in tryptophan-derived metabolites, such as serotonin levels, could impair mood behavior, while changes in melatonin could impair sleep, both parameters affected in aging and neurodegeneration. We detected decreased serotonin levels in the serum of SIRT6 KO mice (Figure 3A), which is consistent with the effect of SIRT6 knockout in HeLA and SHSY-5Y cells (Figure 1F).
Melatonin hormone oscillates in the serum in accordance with dark- light cycles due to transcriptional activation of AANAT. Once expressed, this enzyme becomes the rate-limiting factor in melatonin production and secretion59. The low levels of AANAT and Asmt expression in the brain of brS6KO mice (Figure 2C) suggest an abnormal melatonin secretion cycle. Indeed, wild-type mice showed a normal oscillation of melatonin levels, including an increase during the dark phase, whereas the amplitude of melatonin oscillation in brS6KO mice was significantly smaller and the levels did not increase during the dark phase (Figure 3B). Importantly, we measured the oscillation in gene expression in brS6KO mice brains over a 24-h period and the AANAT gene expression patterns correspond with the oscillations in melatonin levels in both WT and brS6KO mice. The AANAT and melatonin patterns of brS6KO mice are in opposite phases to those of WT. The expression AANAT in brS6KO mice exceeded the expression in controls around the 12-h during light (Figure 3C). In contrast Asmt expression was deregulated over the entire 24-h period while Tph1 was upregulated in most of the time points tested (Figure 3C, Suppl Figure 3A). These findings underscore the role of SIRT6 in modulating serotonin and melatonin secretion through the regulation of rate-limiting enzyme expression.
AANAT mRNA levels in the brain do correlate with melatonin levels, but it needs to be pointed out that C57BL/6 mice, the background strain for our brS6KO model, have a splicing defect in the AANAT60, which results in a reduced melatonin production Therefore, we hypothesized that SIRT6 could influence sleep and circadian patterns not only through melatonin but also through the influence of tryptophan catabolites61.
AhR Activity and Location are Affected by SIRT6 Removal
An interesting TF that is regulated by tryptophan catabolites is AhR, which directly binds and translocate to the nucleus in response to metabolites such as L-kynurenine and kynurenic acid17. AhR affect several pathways in the brain, including neurodegeneration, brain development, sleep patterns and inflammation17–20,23–25,62–65.
AhR, when activated by tryptophan metabolites such as L-kynurenine and kynurenic acid, can translocate to the nucleus and affect and affect its target genes17. Therefore, we decided to test AhR activity in SIRT6 KO models. First, we checked for enrichment of AhR consensus motif and binding sites in genes differentially expressed (DE) in the brains of brsS6KO mice versus brains of wild-type animals, using RNA-seq data. Notably, more than 2000 genes affected by SIRT6 are putative AhR targets, many more than what would be expected by chance (Figure 4A-B). among these DE genes, upregulated gene categories include axon guidance and synapse organization, while genes involved in metabolic and mitochondria functions are overall downregulated (Figure 4C). Moreover, out of the 21 differentially expressed genes involved in the circadian rhythm, 19 (90%) are predicted AhR targets (Figure 4D).
Importantly, out of the DE genes predicted to be AhR targets are the rate-limiting enzymes IDO1,2 and Kynu but not TDO2 (Suppl. Figure 4A). These findings suggest that the effects of SIRT6 deficiency on the brain are, at least in part, mediated via AhR. Furthermore, the AhR mRNA levels were higher in the brains of brS6KO mice versus the brains of WT animals (Figure 4E), which is consistent with the enrichment of SIRT6 binding to AhR promotor region and gene (Suppl. Figure 4B). However, despite the higher levels of AhR mRNA on brS6KO vs wild-type mice (Figure 4E), recruitment of AhR to chromatin was significantly reduced in the brains of brS6KO mice (Figure 4F), while no significant difference was observed in the total amount of AhR in the brain (Suppl. Figure 4C). To better understand this discrepancy, we thought that the effect of SIRT6 on AhR could be direct or indirect. 1-direct, by protein-protein interaction or 2-inderect possibly mediated via changes in tryptophan catabolism.
To test the indirect effect, we assessed the levels of AhR and SIRT6 in the nuclei of primary mice neurons from shCtrl or shSIRT6, under different conditions (no treatment, 10 µM kynurenic acid, and 100 µM tryptophan) (Figure (4G-H, Suppl. Figure 4D).
SIRT6 silencing itself (which eliminated about 50% of the SIRT6 signal) did not result in a significant change of the AhR presence in the nuclei, possibly because the silencing effect alone was not strong enough to affect AhR as the KO. However, exposure to kynurenic acid or tryptophan increased the AhR signal in the nuclei of control cells. In the SIRT6-silenced cells, exposure to tryptophan did not result in significant changes in the AhR signal in the nuclei, whereas exposure to kynurenic acid resulted in signal reduction. Suggesting that unlike normal cells where AhR translocated as a response to the metabolites, in SIRT6 deficient cells, AhR is not enriched at the nucleus/chromatin.
To test if SIRT6 possesses a direct role in AhR residence in the nucleus we measured direct interaction between SIRT6 and AhR, by performing co-immunoprecipitation experiments in chromatin extracts using Flag-SIRT6. AhR co-precipitated with SIRT6 (Suppl. Figure 4E). This suggests a direct in vivo interaction between the two proteins, indicating that in the absence of SIRT6, AhR has impaired chromatin binding, as previously demonstrated (Figure. 4F). Moreover, we analyzed SIRT6 binding to AhR target genes by ChIP-Seq in SHSY5Y cells and found that SIRT6 binds in this cell line 375 out of 846 AhR-ARNT (Aryl hydrocarbon receptor nuclear translocator) target genes as predicted by TF-ChEA analysis and are hence directly regulated by SIRT6 (Suppl. table 4F). This colocalization of SIRT6 and AhR at hundreds of target genes implies that in the absence of SIRT6, direct interaction and co-regulation of shared genes would be impaired. These results suggest that SIRT6 modulates AhR activity through several mechanisms, including transcriptional regulation, control of tryptophan catabolism, and direct interaction (see graphic summary Figure 4I), leading to disrupted gene expression.
Lack of SIRT6 Impairs the Circadian Clock in the Brain
AhR is known to interact with the circadian clock components, and its over-activation by its ligands impairs the circadian regulation20,66. Furthermore, it is known that SIRT6 possesses a role in circadian clock regulation in peripheral tissue67,68 but it was never demonstrated in the brain, the pacemaker of the whole body circadian machinery. Since sleep quality decays with age and neurodegeneration this could be affected by SIRT6-AhR axis. Therefore, we analyzed the transcriptomic profile of the circadian clock pathway in brains of brS6KO mice, and they are significantly different from the corresponding profile of wild-type animals (Figure 5A-B). We explored the expression of the negative regulators of the circadian machinery, Cry1, PER1, and PER2, in 6-hour intervals (total of 24h with 12:12 light-dark cycle). The mRNA levels of PER1 and PER2 (but not Cry1) were consistently higher in brS6KO animals; this discrepancy was not observed in the liver, where SIRT6 is present (Figure 5C).
Since metabolic genes respond to the circadian clock machinery69,70, we measured the oscillation in tryptophan-related gene expression in the brain over a 24-h period. We found that the levels of IDO2, TDO2 and Ddc (Figure 5D, Suppl. Figure 5A), but not IDO1 (Suppl. Figure 5A) were elevated at all time points, showing an impairment in the circadian regulation of gene expression.
Moreover, testing relative abundance of BMAL1 and CLOCK, the core transcription activators of the circadian clock machinery, revealed no significant differences in the total brain extract of brS6KO mice and controls (Suppl. Figure 5B). However, these two proteins were significantly less abundant in the chromatin extract of brS6KO mice (Fig. 5E), which suggests a role of SIRT6 in their chromatin recruitment.
A ChIP-qPCR/seq analysis of PER1, Cry1, and CLOCK promoters shows a significant enrichment of SIRT6 binding, compared to the negative control, suggesting that SIRT6 is directly involved in the regulation of their transcription also in the brain (Suppl. Figure 5B-C).
These findings highlight the role of SIRT6 in regulating circadian clock machinery in the brain, both through AhR regulation and independently, affecting also the metabolite production. This underscores its importance in maintaining proper circadian rhythms, which are crucial for sleep quality – a property that is fundamentally impaired in aging and neurodegeneration.
Brain SIRT6 KO Mice (brS6KO) Have Impaired Circadian Patterns
Sleep cycle disruption, and fragmented sleep are common features of aging and neurodegeneration71–75 often related to decreased melatonin secretion and loss of the robustness of the circadian machinery in aging9,10,76,77. Given the shifts in gene expression observed in the brains of brS6KO mice, we speculated that these changes might also lead to a behavioral shift.
We monitored the activity of WT and brS6KO mice during the dark-light cycles for 21 days in regular dark-light cycles (12:12) Compared with wild-type animals, brS6KO exhibited greater spontaneous activity in the resting (light) hours, as well as shorter sleep episodes (Figure 5F, Suppl. Figure 5E) suggesting decline in sleep quality. To test their response to a disruption of the dark-light cycle, we then kept them in constant darkness for 3 weeks (dark stress), which was followed by a 4-week recovery period, spent in the normal dark-light cycle. After switching from the normal cycle to the period of dark stress, wild-type, and brS6KO mice began progressively shifting the periods of high activity (mice are nocturnal animals) into the hours previously spent resting. However, the effect was more pronounced in brS6KO mice, which underwent a full cycle shift, almost reaching by three weeks the time point 0 (Figure 5G). This progressive shift in activity throughout the dark stress period was linear; its slope (i.e., duration of additional, high-activity period per day of total darkness, see Suppl. Figure 5F for calculation explanation) was greater in brS6KO mice than in controls (Figure 5H), and so was the average shift in waking hours (12 h vs. 7 h; Figure 5I). Overall, lack of SIRT6 not only affects circadian behavior in mice but also impaired sleep quality, with spontaneous awakening, shorter sleep episodes during sleep hours, and less resistance to circadian stress.
A SIRT6 KO Drosophila Melanogaster model for neurodegeneration
Mouse models tend to have different metabolic behavior than humans, and in many cases, changes observed in mouse models are not conserved in humans; moreover, the mutation in our model regarding melatonin production could bias some of our findings; therefore, we decided to develop a drosophila model for SIRT6-KO which allowed faster interventions and could suggest that if the pathways are conserved from drosophila to mouse, the probability of these changes to be conserved to a human would be higher. Recently two SIRT6-deficient Drosophila strains were developed, each showing reduced longevity and metabolic impairment and showing a viable model78,79.
First, to validate our own SIRT6-KO Drosophila model, we analyzed the mutation sequence using BLAST. A stop codon was generated after 30 amino acids, suggesting a nonsense-mediated mRNA decay or the formation of a truncated protein (data not shown). Since there are no available antibodies for dSIRT6, we cloned this transcript into a mammalian vector with a Flag tag on the N-terminus and transfected SHSY5Y SIRT6KO cells to test whether the mutated sequence could generate a different SIRT6 isoform. Only cells transfected with full-length SIRT6-Flag showed protein production, while the expression of our mutated Drosophila sequence could not be detected. In addition, transfection of SIRT6KO cells with the full-length SIRT6-Flag reduced the acetylation of histone H3 on lysine residues 56 and 9 (known targets of SIRT6 activity), while transfection with the Drosophila mutant had no effect (Suppl. Figure 6A). Importantly, these histone acetylation patterns were also observed in fly protein extracts (Suppl. Figure 6B). Thus, our novel SIRT6-KO drosophila melanogaster model follows the expected epigenetic patterns of SIRT6 deficiency.
Behavioral and Metabolic Impairments in SIRT6 KO Drosophila Mimic Those in brS6KO Mice
To test whether this model presents a degenerative phenotype, we performed a behavioral test commonly used to assess aging and neurodegeneration; negative geotaxis ability (climbing) to determine neuronal/motor impairment. SIRT6 KO flies showed an impairment in climbing ability versus control flies as early as seven days post eclosion, and this discrepancy increased with age (Figure 6A, Suppl Figure.7). This suggests an accelerated onset of aging, which has also been observed in SIRT6 KO mice80. In addition, in the brains of SIRT6 KO Drosophila, there was an increase in the relative amounts of H3K9ac (a SIRT6 target, Suppl. Figure 6B) and the phosphorylated H2AX and 53BP1 (indicators of DNA damage), versus control animals (Figure 6B). This is similar to brS6KO mice, which also exhibit reduced deacetylation of target proteins and increased DNA damage signaling38.
Moreover, using the Behnke et al. protocol81, we observed a greater number of vacuoles and a greater average vacuole area in the brains of SIRT6KO Drosophila versus control flies, which indicates a higher degree of neurodegeneration (Figure 6C-D, Suppl. Figure 6C).
Therefore, we developed and validated a new Drosophila model for SIRT6 deficiency that shows epigenetic alterations, DNA damage, accelerated neurodegeneration, and behavioral impairment.
Finally, our metabolomic findings indicate that tryptophan metabolism in SIRT6 mutant flies is shifted in ways similar to those observed in brS6KO mice. In drosophila, the tryptophan-metabolizing enzymes TDO2 and KMO are encoded by Vermillion and Cinnabar genes, respectively. The expression of Vermillion was significantly higher in the heads of SIRT6 KO Drosophila than in control flies, whereas the effect was the opposite for Cinnabar (Figure 6E).
SIRT6 KO flies also have higher levels of tryptophan, kynurenic acid, and kynurenine versus controls and lower levels of xanthurenic acid and melatonin (Figure 6F, Suppl. Figure 6D-E), which is consistent with the changes in expression of rate-limiting enzymes TDO2 and KMO, and similar to the metabolomics findings obtained in SH-SY5Y, HeLa, and ES KO cells (Figure 1C-D, F; Suppl. Figure 1A).
SIRT6 KO-related changes in metabolite levels were observed in both sexes, except in the case of dopamine, for which significantly lower levels in SIRT KO vs control flies were observed in males only (Suppl. Figure 6D).
We also analyzed Drosophila RNA sequences published by Taylor et al. 79 and found an overall reduction of tryptophan transporter Slc7a5 mRNA in flies overexpressing SIRT6 versus control animals (Figure 6G). In addition, the abundance of tryptophan transporter increased with age in wild-type flies only (Figure 6G), corresponding with our findings in mice (Figure 1E).
Finally, we observed a significant decrease in Cry expression and no significant change in PER in SIRT6 KO Drosophila brains versus control flies (Suppl. Figure 6E).
Overall, our results suggest a conserved role for SIRT6 in regulating epigenetic changes, DNA repair, tryptophan catabolism, the circadian clock and preventing neurodegeneration.
TDO2 Inhibition Improves Climbing Ability in SIRT6KO Drosophila Flies
The kynurenine pathway has been identified previously as a potential target for the treatment of aging and neurodegeneration1–4,6,82,83. In C. elegans, TDO depletion was shown to extend lifespan and delay age-related decline in protein homeostasis2.In Drosophila models of AD and PD, pharmacological inhibition of TDO improved disease-specific pathology, as well as the climbing ability83.
We speculated that pharmacological inhibition of TDO2 will prevent byproducts accumulation and would improve climbing ability in SIRT6 KO Drosophila. Therefore, we treated our SIRT6 KO Drosophila flies with 100 µM 680C91, an inhibitor of TDO2 and vehicle only (DMSO), for 21 days and did the same with wild-type flies (Figure 7A). At 14 and 21 days, climbing ability of SIRT6 KO flies, with or without TDO2 inhibition, remained significantly inferior to the performance of their wild-type counterparts (Figure 7B). However, we also observed a significant improvement in TDO inhibitor - treated SIRT6 KO flies versus those treated with vehicle only: in males (but not females) at day 14 and in females (but not males) at day 21 (Figure 7B, Suppl. Table 2.1).
In addition, the average vacuolar area in the brains of SIRT6KO flies was reduced by more than 50% with TDO2 inhibitor treatment (Figure 6D). However, the average number of vacuoles per brain more than doubled (Suppl. Figure 6C). This suggests that new vacuoles are forming in SIRT6-deficient brains, and that TDO2 inhibition prevents their development into larger structures.
In contrast, treatment with melatonin did not improve climbing activity in male SIRT6 KO flies and even worsened it in females (Figure 7C, Suppl. Table 2.2), suggesting is not only the lack of melatonin but rather the metabolic imbalance that causes neurodegenerative phenotype.
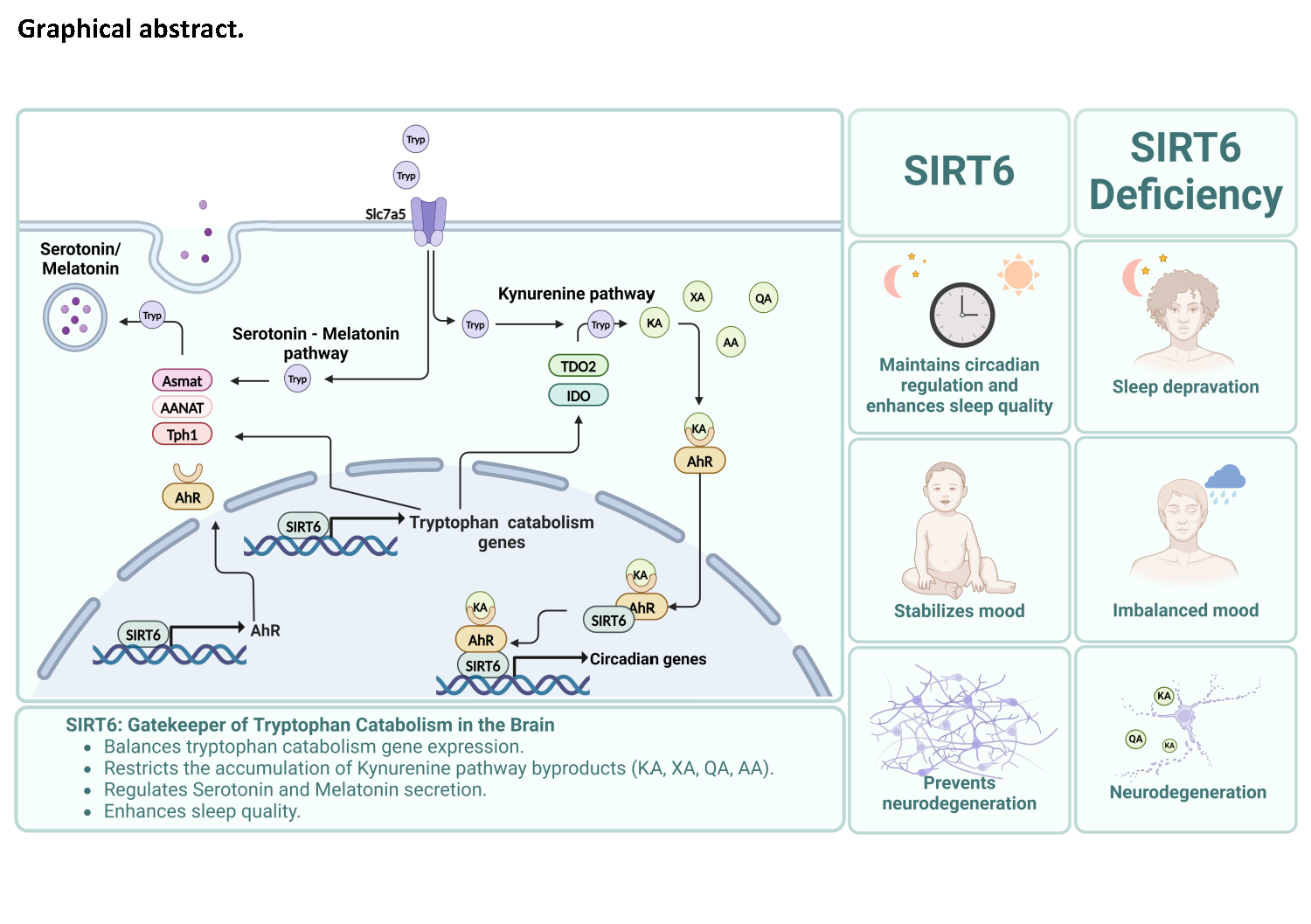
Overall, we showed that SIRT6 impairment leads to changes in tryptophan catabolism, redirecting most of it to the kynurenine pathway, increasing the levels of neurotoxic byproduct and reduction of the levels of serotonin and melatonin in the organism. Many of the metabolites are affecting AhR, leading to changes in gene expression in key metabolic and sleep related pathways. In addition, we established a new Drosophila model that presents neurodegenerative phenotype, which can be alleviated by TDO2 inhibition, by allowing the proper usage of tryptophan. Overall, SIRT6 is a conserved regulator of tryptophan catabolism in the absence of SIRT6, tryptophan re-direction can accelerate the neurodegenerative phenotype through the accumulation of neurotoxic metabolites, and lack of essential signals such as melatonin and serotonin, affecting sleep and behavior (see summary Figure 8).
{kind=link}