Synoeca-MP peptide and chlorhexidine digluconate preparation
Synoeca-MP peptide (INWLKLGKKIIASL-NH2) was synthesized in solid-phase using F-moc methodology, purified (purity > 95%), lyophilized and stored by AminoTech (São Paulo, Brazil) [18]. To confirm molecular mass and purity, Matrix Assisted Laser Desorption Ionization - Time of Flight (MALDI-ToF) mass spectrometry was used (Supplementary Fig. 1). The peptide was dissolved in ultrapure water and stored at -20°C until use. Chlorhexidine was manipulated for stock solution (20% concentrated) (Via Magistral, Brasília, Distrito Federal, Brazil) and dissolved in ultrapure water [19].
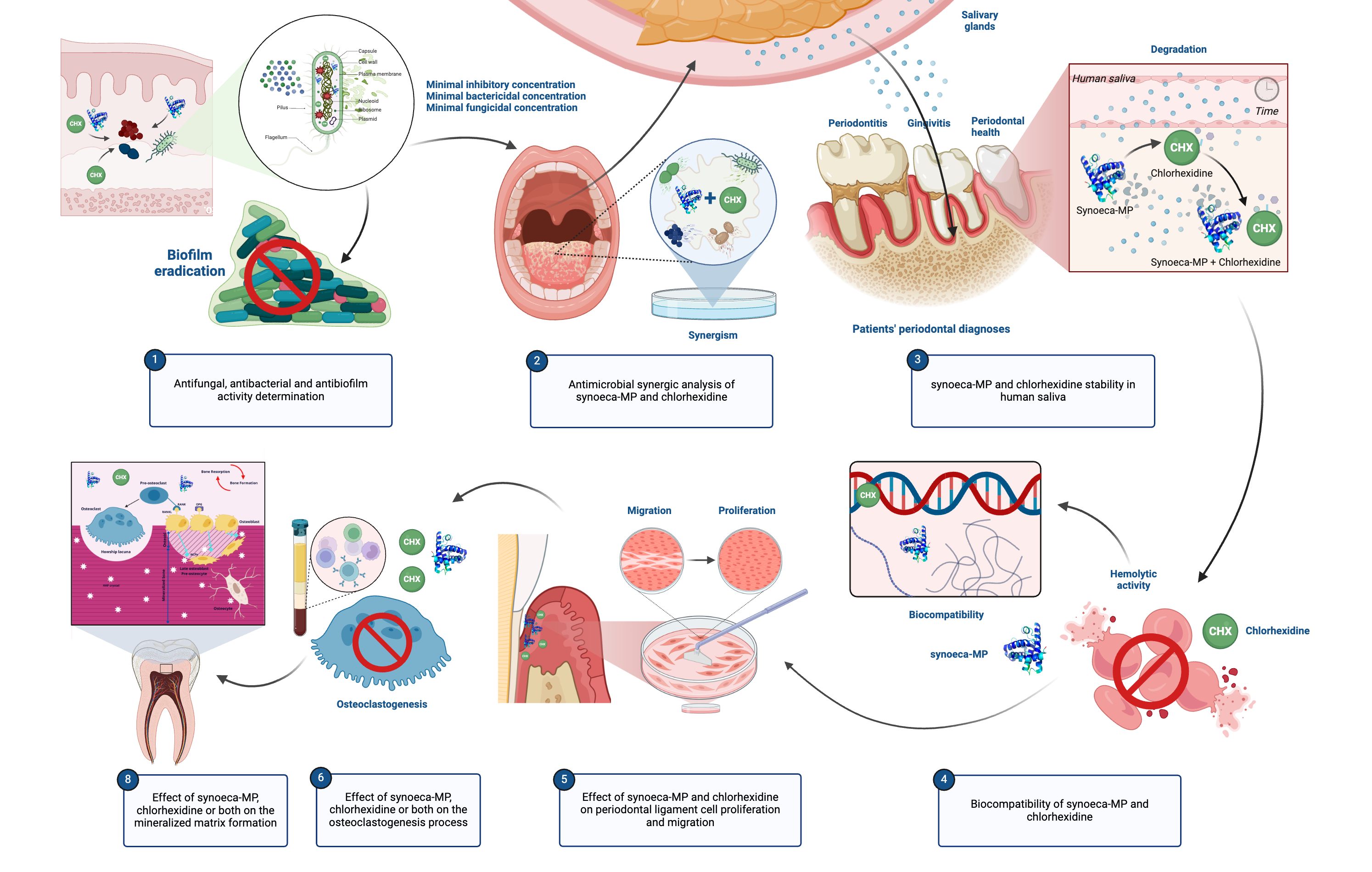
Determination of minimal inhibitory concentration (MIC), minimal bactericidal concentration (MBC) and antibiofilm concentration
Bacteriostatic and bactericidal concentrations were determined according to Clinical & Laboratory Standards Institute guidelines, with some adaptations. Analyses were performed to determine their effectiveness against P. aeruginosa (ATCC27853), E. faecalis (ATCC 19433) and S. aureus (ATCC 25923). Growth curves from each microorganism were previously determined to use its log phase during assays. Antibacterial assays were performed with Luria-Bertani media (Invitrogen, Waltham, Massachusetts, USA) with 5x105 CFU.mL-1 in a 96-well plate for 18h at 37 ºC incubated in a microplate reader with readings at 600 nm absorbance under medium agitation (BioTek PowerWave HT, Winooski, Vermont, USA). Ampicillin (50 µg.mL-1 – Sigma-Aldrich, San Luis, Missouri, USA) was used as a negative control for antibacterial assays. Chlorhexidine (Via Magistral) was tested up to 512 µg.mL-1 (1013 µM) and MIC was determined by no bacteria growth. After MIC determination, each sample was incubated in an agar plate to assess the presence of viable microorganisms for the definition of MBC. Then, antibiofilm properties of synoeca-MP and chlorhexidine were assessed against preformed biofilms of P. aeruginosa and S. aureus. Bacterium inoculum was diluted in a BM2 minimum medium, consisting of 62 mM potassium phosphate buffer (pH 7.0 - VETEC, Duque de Caxias, Rio de Janeiro, Brazil), 7 mM (NH4)2SO4 (VETEC), 2 mM MgSO4 mM (Sigma-Aldrich), 10 µM FeSO4 (VETEC), and 0.5% glucose (Sigma-Aldrich), so that the final concentration reached was 1/100 v/v per well [20]. Bacteria were plated and kept for 24h at 37°C. Controls were represented by P. aeruginosa and S. aureus bacteria (1/100 v/v) as negative control, ciprofloxacin (Sigma-Aldrich) and the BM2 medium [62 mM potassium phosphate buffer (pH 7.0 - VETEC), 7 mM (NH4)2SO4 (VETEC), 2 mM MgSO4 mM (Sigma-Aldrich), 10 µM FeSO4 (VETEC), and 0.5% glucose (Sigma-Aldrich)], as positive control. After 24h, planktonic bacteria were discarded and plates were washed twice with 1x PBS. Serial sample dilutions were performed and added to the preformed biofilm microplates, for 24h. Subsequently, to assess the biofilm’s viability, the MTT assay (Sigma-Aldrich) was performed, according to the manufacturer's standards [21].
Determination of minimal inhibitory concentration (MIC), minimal fungicidal concentration (MFC) and antibiofilm concentration of Candida albicans
Antifungal analyses were performed against C. albicans (ATCC 10231). Experimental assays were performed with RPMI-1640 media (Sigma-Aldrich) with 0.165 mol.L-1 MOPS with 2.5x103 CFU.mL-1 in a 96-well plate for 48h at 37 ºC in a microplate reader under medium agitation (BioTek PowerWave HT). Amphotericin B (10 µg.mL-1 – Sigma-Aldrich) was used as a negative control for antifungal assays. Chlorhexidine (Via Magistral) was tested up to 512 µg.mL-1 (1013 µM) and MIC was determined by no fungal growth. After MIC determination, the sample was incubated in an agar medium for determination of MFC. Fungal antibiofilm properties of synoeca-MP and chlorhexidine were assessed against preformed biofilms of C. albicans. Then, cells were centrifuged, after 48 h growth, at 3000 g, 5 min, at 4 ºC, washed twice with sterile PBS and then resuspended in BM2 media [62 mM potassium phosphate buffer (pH 7.0 - VETEC), 7 mM (NH4)2SO4 (VETEC), 2 mM MgSO4 mM (Sigma-Aldrich), 10 µM FeSO4 (VETEC), and 0.5% glucose (Sigma-Aldrich)]. Cells were seeded at 1x107 CFU per well in 96-well plates and incubated for 48h at 37 ºC. Antimicrobial agents were added to plates and incubated for 24h more. All agents were tested according to the following concentrations: chlorhexidine up to 128 µg.mL-1 (253 µM) and synoeca-MP up to 128 µg.mL-1 (80 µM). Amphotericin B (Sigma-Aldrich) at 16 µg.mL-1 was used as a positive control. The effect on biofilms was evaluated by XTT cell viability kit assays (Biotium, Fremont, California, USA) and plates were read at 470 nm absorbance [22].
Synergism assay
After the determination of minimum inhibitory concentrations, the synergism assay was assessed for the combination of chlorhexidine and synoeca-MP using the microdilution growth inhibition assay protocol, according to the Clinical and Laboratory Standards Institute (CLSI) [23]. Thus, concentrations below its MICs were tested according to the checkerboard method with adaptations [24]. Assays were performed against P. aeruginosa, S. aureus and C. albicans as previously described to determine MIC. The fractional inhibitory concentration (FIC) index determined synergistic interactions according to the following calculation: [(MIC of 1 combined to 2) / (MIC of 1)] + [(MIC of 2 combined to 1) / (MIC of 2)]. Values < 0.5 were considered as a synergic interaction.
Degradation/integrity of synoeca-MP and chlorhexidine in human saliva
The protocol of this study was approved by the Human Research Ethics Committee (number 90666218.2.0000.0029). Saliva was collected from control patients (without gingival and/or periodontal diseases; n = 7), patients diagnosed with gingivitis (n = 6), and periodontitis (n = 17), prior to clinical dental care, at the graduate dentistry clinic of Universidade Católica de Brasília. Patient data for those with periodontal disease were obtained from medical records, and saliva samples were taken between 8 and 11 am. Unstimulated saliva was collected for 5 min [25]. Subsequently, 80.26 µM (40.565 µg/mL) of chlorhexidine, 80.26 µM (128 µg/mL) of synoeca-MP, or the same concentration of chlorhexidine and synoeca-MP together were exposed to saliva; the control group was represented by no treatment exposed to saliva control group. To assess the integrity of synoeca-MP and chlorhexidine in saliva, molecular mass, and purity were evaluated using the autoflex speed spectrometer mass spectrometry technique (Bruker Daltonics, Billerica, Massachusetts, USA), using the reflected and positive operating method, adjusted for 400 to 3500 Da, with external calibration [26]. Synoeca-MP and chlorhexidine were diluted in ultrapure water and deposited on the plate (AnchorchipVar-384 – Bruker Daltonics) together with the matrix in triplicate, awaiting complete crystallization at room temperature after 0, 30, 60, 90, 120, 150, 180, 210 and 240 min. Between the experimental periods, samples were kept at 37°C, to mimic saliva conditions in the body environment. Then, the correlation between the time of degradation of the peptide, chlorhexidine, and their association in the saliva of patients with the different periodontal diagnoses was analyzed.
Periodontal ligament cell culture
Healthy third molars extracted from patients aged 18 to 30 years were used to obtain periodontal ligament cells. Ethics registration and approval had been obtained from the Human Research Ethics Committee of the Catholic University of Brasília (CAAE: 90666218.2.0000.0029) and all donors signed the understanding and written consent. Right after extraction, extracted teeth were placed in a sterile tube containing Dulbecco Modified Eagle Medium (DMEM; Gibco, Grand Island, New York, USA) without FBS, associated with collagenase type 1 (3 mg.mL-1) and dispase (4 mg.mL-1), for 1 h at 37 ºC [28]. After this period, tooth roots were shaved, and tissue structures of the periodontal ligament adhered to the roots were collected in 6-well cell culture plates (Costar Corp., Cambridge, Massachusetts, USA). Cells were cultured in supplemented DMEM culture medium with 10% FBS (Gibco), 1% MEM amino acid solution (Gibco), 0.05% gentamicin (Gibco), 1% L-glutamine (Gibco), 1% penicillin / streptomycin (1000 U.mL-1) (Gibco) and kept at 5% CO2, 37 ºC and at 95% humidity [29]. Every three days, the culture medium was changed, until reaching the number of cells required for the experiment.
Hemolytic assay
The hemolytic activity of synoeca-MP and chlorhexidine was based on the methodology described by Bignami, with adaptations [27]. Thus, a suspension with 1% cells in PBS was prepared and centrifuged twice (1000 x g, for 2 min). Subsequently, 350 µL of the 1% erythrocyte solution was used to test different concentrations of synoeca-MP and chlorhexidine. Controls were represented by saline solution (0% hemolysis, negative control) and triton X-100 (Sigma-Aldrich) at 0.1% (100% hemolysis, positive control). After 1h, tubes were centrifuged and 100 µL of the supernatant was collected to determine the optical density in a microplate reader at 406 nm.
MTT assay
Human peripheral blood mononuclear cells (PBMCs), periodontal ligament cells, and SaOs-2 viability were evaluated according to the experimental groups and incubation period of each proposed assay. The positive control was represented by cells in culture medium (100% of cell viability) and the negative control was represented by cells in lysis solution − 10 mM Tris (Sigma-Aldrich), pH 7.4, 1 mM EDTA (Sigma-Aldrich) and 0.1% triton X-100 (Sigma-Aldrich), representing 0% cell viability. At the end of the incubation period, the MTT colorimetric assay (Sigma-Aldrich) was used [21].
Synoeca-MP and chlorhexidine on cell proliferation and migration assay
Cells were plated in 96-well plates (Kasvi, São José dos Pinhais, Paraná, Brazil) and after 24h, synoeca-MP and chlorhexidine stimuli were added. Then, after 24 h, 48 h and 72 h, cell viability was determined by MTT assay. Cell proliferative potential in the presence of tested substances was performed using the Tripan Blue exclusion technique, after 0h, 24h, and 48h of cell incubation [30]. Confluent periodontal ligament cell cultures were obtained, and their trypsinization and cell resuspension assays were performed in DMEM medium (Gibco) without FBS (same conditions as those used in cell migration assay). Periodontal ligament cells (1x105 cells) were added in 1 mL DMEM (Gibco) without FBS to 24-well plates. Experimental groups contained the lowest antimicrobial synergistic concentration of synoeca-MP and chlorhexidine, associated or alone. Then, after completing the experimental incubation, cells were resuspended and the solution was added to 0.4% trypan blue dye (Sigma-Aldrich), for 1 minute. Cells were counted immediately using a Neubauer chamber (Brand GmbH, Wertheim, Baden-Württemberg, Germany). To assess the effect of synoeca-MP, chlorhexidine and their association on cellular proliferation, a scratch assay was performed [31]. Cells from the periodontal ligament were kept confluent, and through the accomplishment of 3 markings, cell adhesion in the plate was disrupted. After that, the plate was washed with PBS, followed by the addition of supplemented DMEM culture medium, with the addition of all tested substances. Then, after 0 h, 24 h and 48 h, photographs were taken in an inverted electron microscope, with a 10x increase, in order to verify the migratory activity in these periods [32]. The measurement of cell migration was performed through the analysis of photographs by counting cells using Image J software [33].
Evaluation of synoeca-MP and chlorhexidine on osteoclastogenesis process
Whole blood (about 4 mL) was collected from healthy volunteers. PBMCs were isolated by density gradient centrifugation using Ficoll-Paque (Sigma-Aldrich), following the manufacturer's recommendations. Then, the cell pellet was resuspended in 10 mL of supplemented DMEM (Gibco) and 25 ng.mL-1 of macrophage colony-stimulating factor recombinant (M-CSF; Peprotech, Rocky Hill, Connecticut, USA) [34, 35]. After 72h, non-adherent cells were removed, and the adherent cells were plated in 96-well plates (Kasvi). After 24h, synoeca-MP and chlorhexidine stimuli were added. Cellular viability was assessed after 14 days. PBMCs (M-CSF-dependent macrophages) were incubated at 1.6x104 cells per well, in 96-well culture plates (Kasvi) with supplemented DMEM (Gibco). Cultures were subjected to stimuli, such as 10 ng.mL-1 of the soluble receptor activator of nuclear factor-κB ligand (sRANKL; Peprotech) and synoeca-MP at its synergistic concentrations with chlorhexidine, associated or alone, for 14 days [36]. After the fourteenth day, the PBMCs were subjected to tartrate-resistant acid phosphatase (TRAP) staining for later counting of the number of differentiated osteoclasts and the number of nuclei per osteoclast in an inverted microscope. For TRAP staining, the acid phosphatase kit, leukocyte (Sigma-Aldrich) was used, following the manufacturer's recommendations. Osteoclasts were considered TRAP-positive cells (red/orange color), with more than three nuclei inside them [37].
Evaluation of synoeca-MP and chlorhexidine on mineral matrix formation
To evaluate mineral matrix deposition effects, periodontal ligament cells, and SaOs-2 (derived from human osteosarcoma) cells were used [38]. Periodontal ligament cells and SaOs-2 cells were cultured at a concentration of 1.6x104 cells per well, in 6-well culture plates (Kasvi) for 21 days, in supplemented DMEM medium (Gibco), with osteogenic conditions (50 µg.mL-1 ascorbic acid, 100 nM dexamethasone, 1 mM β-glycerophosphate; Sigma-Aldrich), for 21 days. Medium and all stimuli were changed every 3 days. Cultures were subjected to synoeca-MP and chlorhexidine stimuli in synergistic concentrations, after 24h [39, 40]. After the incubation period, the concentration of alkaline phosphatase (ALP) and the formation of the mineral matrix were assessed.
Alkaline phosphatase. Determination of alkaline phosphatase (ALP) concentration in periodontal ligament and SaOs-2 cell cultures was assessed after 21 days under osteogenic conditions. ALP was measured by the colorimetric method of paranitrophenol, using the Sigmafast p-Nitrophenyl phosphate kit (Sigma-Aldrich). For the ALP assay, the cells were washed with PBS and incubated in 0.05% Triton X-100 for 20 minutes at room temperature, with constant agitation. Cells were transferred, vortexed for 20 seconds, centrifuged for 15 min at 4°C at 2500 RPM, and kept on ice for 20 minutes. Aliquots of cell lysate were incubated with p-Nitrophenol phosphate (p-NF) substrate at 37 ºC for 30 minutes. The reaction was stopped by adding 5 µL 1N NaOH and the absorbance was measured at 405 nm using the microplate reader (Bio-Tek PowerWave HT). A standard p-NF curve was established to determine enzyme activity. The samples were normalized, and protein quantification was determined by Qubit®ฏ [41].
Mineral matrix formation assay. After 21 days of culture, the formation of the mineral matrix of periodontal ligament cells and SaOs-2 cells was determined by alizarin red staining. For this, plates were fixed with 10% (v / v) formaldehyde (VETEC), at room temperature for 15 min. The cell layer was washed twice with distilled water and 1 mL of alizarin red S dye (40 mM; pH 4.1; Sigma-Aldrich) was added per well. The plate was incubated at room temperature for 20 minutes, under constant agitation. After discarding the unincorporated dye, the cell layer was washed 4 times, with 4 mL of distilled water, with a 5-minute stirring between washes. Then, the distilled water was discarded, and the stained cell layer was evaluated under an inverted microscope (Zeiss, Oberkochen, Baden-Württemberg, Germany). To quantify the stained mineral matrix, 800 µL of acetic acid 10% (v/v) (Dinâmica, Indaiatuba, São Paulo, Brazil) was added to each well, followed by incubation for 30 minutes at room temperature, under constant agitation. The cell layer, together with 10% (v/v) acetic acid (Dinâmica), was then scraped from the bottom of the well, vortexing for 30 seconds, covered with 500 µL of mineral oil (VETEC), heated to 85 ºC for 10 minutes and cooled on ice, for 5 minutes. After complete cooling, the suspension was centrifuged at 20,000 x g for 15 minutes and 500 µL of the supernatant transferred to a new tube. For acid neutralization, 200 µL of 10% (v / v) ammonium hydroxide (VETEC) was added. Aliquots (150 µL) of the supernatant were arranged in triplicate in a 96-well plate (Kasvi) for reading in a microplate reader (Bio-Tek PowerWave HT), at 405 nm [41].
Statistical analysis
All experiments were performed in technical and biological triplicates. Data normality was tested by the Shapiro Wilk test, and parametric variables are described as mean and standard deviation or mean and standard error of the mean. Statistically significant differences were considered when p < 0.05. To measure the statistical differences in the continuous variables, the two-way ANOVA was used followed by the Bonferroni post hoc test. The Kaplan-Meier curves were used to show molecule degradation over time (synoeca-MP, chlorhexidine and synoeca-MP plus chlorhexidine), and statistical differences were assessed by Log-Rank (Mantel-Cox) test. Statistical analysis was performed using the GraphPad Prism 5 software (California, USA).


{kind=link}