Chemistry:
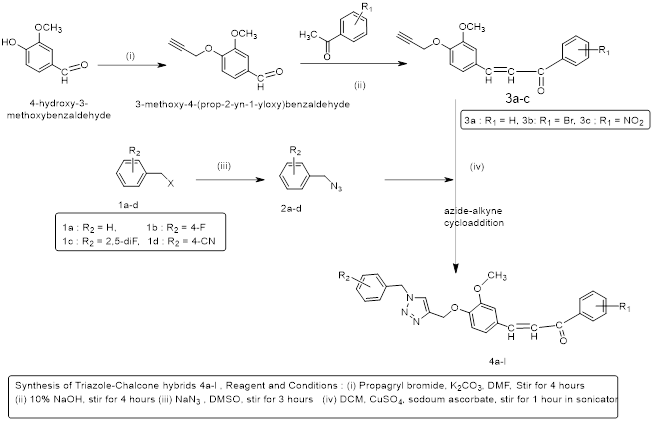
The synthesis of chalcone-triazole is being depicted in scheme-I that involves four steps. In the first step benzyl azide (1a-d) were prepared from benzyl chloride and sodium azide, different substituents on phenyl ring of benzyl chloride are used so that differently substituted triazole could be produced in the later stage. In another step, vanillin was functionalized at O-atom with propargyl group (compound 2) after reacting with propargyl chloride in mildly basic medium. The reaction proceeded very efficiently and gave single product without the formation of side products. The polar solvent is used for the reaction that is DMF but for the formation of pure crystals complete removal of solvent is necessary. Next step involved the formation of chalcone (3a-c), in that propargyl functionalized vanillin (2) was condensed with acetophenones through the aldehydic group via Claisen-Schmidt reaction. The product obtained in this step is not so pure therefore, column chromatography was done to get pure compound. Consequently, the yield of the product was less. The yellow crystals so obtained were recrystallized with ethanol.
The last step is the formation of triazole-chalcone hybrid (4a-l) in that cyclization of propargyl group of functionalized chalcone (3a-c) was done with the aid of Click chemistry tools. In this reaction, azide group of benzyl azides (1a-d) add onto the triple bond of propargyl group of chalcones. The mechanism of the reaction is (3 + 2) cycloaddition. The reaction is catalyzed by CuSO4. The reaction mixture was stirred in a sonicator. The rules of green chemistry are followed in this step.
IR analysis:
The presence of a characteristic band at around 2092.83 cm− 1 indicated the presence of Ν = N = N stretching (in case of benzyl azides) in the IR spectra of molecules (1a-d) and it also showed absorption bands at 1647.26cm− 1 attributing to C = C stretching in aromatics. The IR spectra of compound 2 showed absorption band at ν = 3244.38 cm− 1 due to presence of C-H stretching supporting the attachment of propargyl group with vanillin, apart from this the absence of O-H stretching peak supported the replacement of H of hydroxyl group with propargyl group. Apart of these, absorption peaks at 1120.68 cm− 1 due to C-O stretching, 1689.70 cm− 1 (CHO stretching), p-substituted stretching at 810.13 cm− 1, and o-substituted stretching = 777.34 cm− 1 were also obtained.
The IR spectra of compounds 3(a-c) supported the formation of chalcones with characteristic peaks of C = C stretching at around at 1530.05 cm− 1, and an intense peak of C = O stretching in the region of 1600–1650 cm− 1, thereby supporting the presence of α, β-unsaturated carbonyl functional group. Two distinguishing peaks at around 3200 cm− 1 and 2100 cm− 1 due to C-H stretching and CC stretching for alkynes respectively, but these peaks are absent in the spectra of triazoles 4(a-j). In the spectra of triazoles there was a peak around 3150 cm-1 due to = C-H str of triazole ring and another peak at 1010 cm− 1 due to (Ν = N = N) ring.
NMR analysis:
The 1H-NMR of compounds 1(a-d) signals at ∂ 7.24–7.56 ppm for aromatic protons and singlet for methylene proton in the region of 2.50–2.55 ppm. The spectra of compound 2 showed characteristic signal for H-CC for propargyl group at δ 2.58 ppm and at δ 4.86 for 2 protons of -OCH2. There was no signal for proton for O-H thereby confirming the attachment of propargyl group. In 1H-NMR spectra of 3 (a-c), the signal for -CHO was not present rather signals for = C-H proton became visible in aromatic region thereby confirming the formation of chalcones. In spectra of 4 (a-l) peak for H-CC for propargyl group at δ 2.58 ppm disappeared instead a singlet signal for -C-H appeared at δ 7.18 ppm showing the formation of triazole nucleus.
Biological activity:
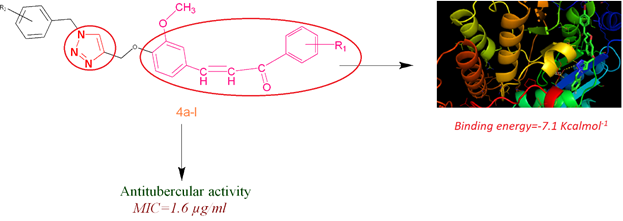
The compound 2–4(h) were evaluated for their Anti-tubercular activity by using H37RV Mycobacterium strain by Alamar blue assay. The Minimum Inhibitor concentration (MIC) is expressed in µg/ml as shown in Table 1. Compound 2 showed activity up to 50µg/ml. Compounds like 3a, 3b, and 3c shows 25 µg/ml, 100 µg/ml and 100 µg/ml. It is worth mentioning that chalcone is unsubstituted it is more potent than the substituted chalcones. Now, when molecular hybrids of these compounds are being formed activity is enhanced. It is seen that unsubstituted triazole exhibit the maximum inhibition at a concentration of 1.6 µg/ml as well when fluorine and bromine both electronegative halogens are attached on triazole ring they also exhibit inhibition upto 1.6 µg/ml. In addition to this, when only fluorine is attached to triazole ring its activity reduced; however, when diflouro is present on ring its inhibition increased upto 1.6 µg/ml. Moreover, substitution of bromine alone showed promising activity up to 6.2 µg/ml but electronegative atom like cyano if present along with bromine that shows antagonistic effect. [] From the results it could be concluded that activity was enhanced on hybridizing the two pharmacophoric unities. These results support the hypothesis that synergistic effect is observed on molecular hybridization. []
Docking Studies:
Molecular docking of biologically active ligands has been performed to study the active binding site in the Mycobacterium protein chain which are available for binding interactions. Moreover, the analysis and interpretation of the binding behavior play a crucial role in potential drug designs and in elucidating fundamentals of biochemical processes. [,]
All the ligands show active binding interaction with the reference protein as indicated by -ve value of binding affinity. All the polar contacts of individual ligands are described in the tabular form with amino acid residue with code. From the data, it is quite evident all the ligands show binding interaction with arginine (ARG) amino acid residue except 4c. This might be helpful in explaining its lower IC50 value as compared to 4a, 4c and 4f. GLN, TYR and SER are other important residue with which ligands binds in similar ways.
Visual presentation of one best binding pose of each ligands is described in figures.
Experimental:
Material: All the chemicals were procured from Sigma Aldrich Ltd. Melting point were recorded in Kofler hot stage apparatus and were uncorrected. 1H NMR were recorded using BRUKER AVANCE II 400 NMR and TMS was used as an internal standard. Coupling constant values are represented in Hz. Infrared spectra were recorded on FTIR 8400S (Shimadzu) and pellet were prepared in KBr. The absorption values were expressed in cm − 1.
Methods:
Synthesis of compounds
1. Synthesis of compound 1-(azidomethyl) benzene
Reaction starts with benzyl bromide, 2, 5- fluorobenzyl bromide, 4-cyanobenzylbromide (5 mmol) and Sodium azide (6 mmol) in DMSO, the reaction mixture was stirred for 4–6 hours at room temperature. Completion of reaction can be confirmed using TLC. After completion of reaction, product was extracted with diethyl ether and the final extract was washed with distilled water to remove off impurities and DMSO if there. Organic layer was dried over Na2SO4 and filtered the mixture. The filtrate was evaporated under rotatory evaporator to get desired compound 1(a-d).
Characterization of compound 1a
The IR spectra of compound 1(a) was recorded just by putting liquid drop on IR plate in the range of 400–4000 cm − 1. It exhibits (aromatic) ν C = C = 1647.26cm− 1, ν Ν = N = N stretching = 2092.83 cm− 1. 1H NMR (400 MHz, DMSO) δ ppm: 7.43–7.33 (m, 5H, C-H Aromatic); 2.50(s, 2H, C-H).
Characterization data of compound 1b
It exhibited (aromatic)ν C = C = 1604.83cm− 1, ν Ν = N = N stretching = 2092.83 cm− 1. 1H NMR (400 MHz, DMSO) δ ppm: 7.43–7.41(d, 2H, C-H Aromatic); 7.24–7.18(d, 2H, C-H Aromatic); 2.55(s, 2H, C-H).
Characterization data o compound 1c
The compound exhibited (aromatic) ν C = C = 1599.04cm− 1, ν Ν = N = N stretching = 2098.62 cm− 1. 1H NMR (400 MHz, DMSO) δ ppm: 7.39–7.28(m, 3H, C-H Aromatic); 2.50(s, 2H, C-H).
Characterization data o compound 1d
The compound exhibited (aromatic) ν C = C = 1610.61cm− 1, ν Ν = N = N stretching = 2094.76 cm− 1. 1H NMR (400 MHz, DMSO) δ ppm: 7.87–7.86(d, 2H, C-H Aromatic); 7.58–7.56(d, 2H, C-H Aromatic); 2.55(s, 2H, C-H).
2. Synthesis of compound 2-methoxy-4-(prop-2-yn-1-yloxy) benzaldehyde (2):
Reaction starts with Vanillin (1.52 g, 0.009 mol), and propargyl bromide (1.36 ml, 0.018 mol) and potassium carbonate K2CO3 (2.76 g, 0.02 mol) were dissolved in 15 ml of DMF. The reaction mixture was stirred for 3–4 hours at room temperature. Completion of reaction was confirmed using TLC technique. After completion of reaction, extraction was done using (Chloroform) CHCl3. Organic layer was separated and washed with excess amount of distilled water to remove off all the impurities and DMF present in the mixture. Final extract was dried over anhydrous Na2SO4. The solvent was evaporated under rotatory evaporator to get compound 2. And recrystallization can be done in ethanol.
Characterization data of compound 2
The IR of compound 2 was done just by putting crystals in IR plate then examined and the spectrum was shown in Fig. 9. The IR peak for νC-H stretching = 3244.38 cm− 1, ν C-O stretching = 1120.68 cm− 1, ν-CHO stretching = 1689.70 cm− 1, ν p-substituted stretching = 810.13 cm− 1, ν o-substituted stretching = 777.34 cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 3.94(s, 3H, -OCH3); 9.87(s, 1H, -CHO); 7.44–7.52 (m, 3H, C-H Aromatic); 4.86(d, 2H, -OCH2); 2.58(t, 1H, C-H).
3. Synthesis of compound (E)-1-(2-methoxy-4-(prop-2-yn-1-yloxy) phenyl)-3-phenylprop-2- sen-1-one (3a-c):
The compound 2(1.36 g, 0.007 mol) and substituted acetophenone, (0.83 ml, 0.007 mol) were dissolved in equimolar NaOH solution in ethanol. The reaction mixture was stirred at room temperature. Progress of reaction was checked using TLC. After the completion of reaction, reaction mixture was poured into chilled distilled water and neutralized the basic solution using HCl. The solid product was filtered which is our desired product 3(a-c).
Characterization data of Compound 3a
The IR analysis of compound 3(a) was done just by putting crystals in IR plate. It exhibited ν C-H stretching = 3292.63 cm-1, ν C = C stretching = 1653.05 cm-1, νC-O stretching = 1138.04 cm− 1, ν o-substituted stretching = 692.47 cm− 1..1H NMR (400 MHz, CDCl3) δ ppm: 7.92(d, 2H, C-H Aromatic); 7.44 (m, 2H, C-H Aromatic); 7.50(m, 1H, C-H Aromatic); 4.74(d, 2H, - OCH2); 3.86(s, 3H, -OCH3); 2.48(t, 1H, C-H); 7.66–7.70(d, 2H, =C-H); 7.31–7.35(d, 2H, =C-H) 7.662(d, 1H, C-H); 7.18(d, 2H, C-H).
Characterization data of Compound 3b
IR spectra indicated ν C-H stretching = 3290.67 cm-1, ν C = C stretching = 1651.12 cm-1, νC-O stretching = 1138.04 cm− 1 ν o-substituted stretching = 698.25 cm-1. 1H NMR (400 MHz, CDCl3) δ ppm: 7.88(d, 2H, C-H Aromatic); 7.64 (m, 2H, C-H Aromatic); 77(m, 1H, C-H Aromatic); 4.88(d, 2H, - OCH2); 3.94(s, 3H, -OCH3); 2.57(t, 1H, C-H); 7.36–7.32(d, 2H, =C-H); 7.24–7.23(d, 2H, =C-H) 7.16(d, 1H, C-H); 7.06(d, 2H, C-H).
Characterization data of Compound 3c
The IR spectrum exhibited ν C-H stretching = 3238.59cm-1, ν C = C stretching = 1662.69 cm-1, ν C-O stretching = 1139.97 cm− 1, ν o-substituted stretching = 644.25 cm-1. 1H NMR (400 MHz, CDCl3) δ ppm: 8.28(d, 2H, C-H Aromatic); 8.06 (m, 2H, C-H Aromatic); 7.73(m, 1H, C-H Aromatic); 4.75(d, 2H, - OCH2); 3.88(s, 3H, -OCH3); 2.49(t, 1H, C-H); 7.69–7.73(d, 2H, =C-H); 7.29–7.25(d, 2H, =C-H) 7.10(d, 1H, C-H); 7.01(d, 2H, C-H).
4.) Synthesis of compound (E)-1-(4-((1-benzyl-1H-1,2,3-triazol-4-yl) methoxy)-2- methoxyphenyl)-3-phenylprop-2-en-1-one (4):
Dissolve compound 3(a-c) (0.1g) and azide 1(a-d) (0.1ml) in 20 ml of dry DCM. After that solutions of anhydrous CuSO4 (0.04 g) and sodium ascorbate (0.25g) in water were added. The solution was stirred at room temperature in sonicator till reaction was completed. Progress of reaction was observed using TLC. After the completion of reaction 50 ml of water was added and extracted with DCM. The organic layer was dried over Na2SO4 and the filtrate was evaporated under rotatory evaporator to get our desired product. And wash the final product with cold ethanol.
Characterization data of Compound 4a
The IR spectra of compound 4(a) exhibited νC = O stretching = 1656.91 cm-1, νC-O stretching = 1134.18 cm− 1, νC = C stretching = 1575.89 cm− 1, νC-N stretching = 1255.70 cm− 1, ν(Ν = N = N) ring = 1010 cm− 1, νC-H aromatic = 2918.40 cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 7.999(d, 2H, C-H); 7.583(d, 1H, C-H); 7.504(d, 2H, C-H); 7.36(m, 4H, C-H); 7.258(t, 3H, C-H);
-
7.18(s, 1H, =C-H); 7.06(s, 1H, =C-H); 7.25(s, 1H, C-H); 7.12(s, 1H, C-H); 5.50(s, 2H, -OCH2);
-
5.31(s, 2H, -CH2); 3.87(s, 3H, -OCH3).
Characterization data of Compound 4b
It exhibited νC = O stretching = 1658.84 cm− 1, νC-O stretching = 1134.18 cm− 1, νC = C stretching = 1585.54 cm− 1, νC-N stretching = 1255.70 cm− 1, ν(Ν = N = N)ring = 981.80cm− 1, νC-H aromatic = 2914.54 cm− 1 ,ν = C-H = 3144.07 cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 8.00(d, 2H, C-H); 7.57(d, 1H, C-H); 7.50(d, 2H, C-H); 7.27–7.24(m, 4H, C-H); 7.143(t, 3H, C-H); .7.18(s, 1H, =C-H); 7.09(s, 1H, =C-H); 7.57(d,1H, C-H); 5.48(s, 2H, -OCH2); 5.31(s, 2H, -CH2); 3.90(s, 3H, -OCH3) .
Characterization data of Compound 4c
IR peaks at ν C = O stretching = 1660.77 cm-1, νC-O stretching = 1130.32 cm-1, νC = C stretching = 1587.47 cm− 1, νC-N stretching = 1244.13 cm-1, ν (Ν = N = N)ring = 985.66cm− 1, νC-H aromatic = 2918.40 cm− 1, ν = C-H = 3140.53 cm− 1. . 1H NMR (400 MHz, CDCl3) were observed.
δ ppm: 8.01(d, 2H, C-H); 7.76(d, 2H, C-H); 7.56(d, 2H, C-H); 7.51(d, 2H, C-H); 7.08(t, 3H, C-H 7.14(s, 1H, =C-H); 7.07(s, 1H, =C-H); 7.25(s, 1H, C-H); 5.48(s, 2H, - OCH2); 5.31(s, 2H, -CH2); 3.94(s, 3H, -OCH3)
Characterization data of Compound 4d
IR spectra showed peaks at νC = O stretching = 1658.84 cm-1, ν C-O stretching = 1132.25 cm-1, νC = C stretching = 1585.54 cm− 1, νC-N stretching = 1255.70 cm-1, ν(Ν = N = N) ring = 981.80cm− 1, ν C-H aromatic = 2914.54 cm− 1, ν = C-H = 3139.46 cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 8.00-7.25(m, 13H, Ar-H); 7.30(s, 1H, =C-H)); 7.14(s, 1H, =C-H)); 5.50(s, 2H, -OCH2); 5.36(s, 2H, -CH2); 3.80(s, 3H, -OCH3).
Characterization data of Compound 4e
The IR of compound exhibited νC = O stretching = 1654.98 cm-1, νC-O stretching = 1132.25 cm-1, νC = C stretching = 1581.68 cm− 1, νC-N stretching = 1257.63 cm-1, ν (Ν = N = N) ring = 1009.09cm− 1, νC-H aromatic = 2918.40cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 7.88–7.20 (m, H, Ar-H); 7.19(s, 1H, =C-H)); 7.12(s, 1H, =C-H)); 5.47(s, 2H, -OCH2); 5.28(s, 2H, -CH2); 3.89(s, 3H, -OCH3).
Characterization data of Compound 4f
The compound exhibited νC = O stretching = 1654.98 cm-1, νC = N = 1654.98, νC-O stretching = 1132.25 cm-1, νC = C stretching = 1585.54 cm− 1, νC-N stretching = 1255.70 cm-1, ν(Ν = N = N)ring = 977.94scm− 1, νC-H aromatic = 2918.40cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 7.99–7.35(m, 13 H, Ar-H); 7.10(s, 1H, =C-H)); 7.08(s, 1H, =C-H)); 5.51(s, 2H, -OCH2); 5.35(s, 2H, -CH2); 3.90(s, 3H, -OCH3).
Characterization data of Compound 4g
IR spectra showed the presence of νC = O stretching = 1654.98 cm-1, νC-O stretching = 1130.32 cm-1, νC = C stretching = 1577.82 cm− 1, νC-N stretching = 1232.55 cm-1, ν(Ν = N = N)ring = 993.37scm− 1, νC-H aromatic = 2918.40cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 8.00-7.35(m, 12 H, Ar-H); 7.10(s, 1H, =C-H)); 7.08(s, 1H, =C-H)); 5.54(s, 2H, -OCH2); 5.32(s, 2H, -CH2); 3.99(s, 3H, -OCH3).
Characterization data of Compound 4h
The compound exhibited νC = O stretching = 1654.98 cm-1, νC-O stretching = 1130.32 cm-1, νC = C stretching = 1579.75 cm− 1, νC-N stretching = 1253.77 cm-1, ν(Ν = N = N) ring = 995.30cm− 1, νC-H aromatic = 2916.47cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 8.00-7.35(m, 13 H, Ar-H); 7.10(s, 1H, =C-H)); 7.08(s, 1H, =C-H)); 5.58(s, 2H, -OCH2); 5.33(s, 2H, -CH2); 3.91(s, 3H, -OCH3).
Characterization data of Compound 4i
The IR spectra exhibited the presence of νC = O stretching = 1656.91 cm-1, νC-O stretching = 1134.18 cm-1, νC = C stretching = 1573.97 cm− 1, νC-N stretching = 1257.63 cm-1, ν(Ν = N = N) ring = 985.66cm− 1, νC-H aromatic = 2918.40cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 8.00-7.35(m, 14 H, Ar-H); 7.35(s, 1H, =C-H)); 7.31(s, 1H, =C-H)); 5.52(s, 2H, -OCH2); 5.28(s, 2H, -CH2); 3.91(s, 3H, -OCH3).
Characterization data of Compound 4j
The IR spectra showed the presence of νC = O stretching = 1680.05 cm-1, νC-O stretching = 1132.25cm-1, νC = C stretching = 1585.54 cm− 1, νC-N stretching = 1257.63 cm-1, ν(Ν = N = N) ring = 997.23cm− 1, νC-H aromatic = 2918.40cm− 1.
Characterization data of Compound 4k
IR analysis showed νC = O stretching = 1678.13 cm-1, νC-O stretching = 1134.18cm-1, νC = C stretching = 1585.54 cm− 1, νC-N stretching = 1257.63 cm-1, ν(Ν = N = N) ring = 999.16cm− 1, νC-H aromatic = 2918.40cm− 1. 1H NMR (400 MHz, CDCl3) δ ppm: 8.00-7.35(m, 13 H, Ar-H); 7.21(s, 1H, =C-H)); 7.12(s, 1H, =C-H)); 5.48(s, 2H, -OCH2); 5.34(s, 2H, -CH2); 3.82(s, 3H, -OCH3).
Characterization data of Compound 4l
The IR analysis gave the presence of νC = O stretching = 1678.13 cm-1, νC-O stretching = 1134.18cm-1, νC = C stretching = 1585.54 cm− 1, νC-N stretching = 1257.63 cm-1, ν(Ν = N = N) ring = 999.16cm− 1, νC-H aromatic = 2918.40cm− 1.
Methods to analyze Anti-tubercular activity:
The anti-TB activity was done using Alamar blue assay. All the synthesized compounds (Scheme-I) µwere tested against Mycobacterium tuberculosis. 100 µl each of sterile water and Middlebrook 7H9 broth was added in each well of 96 well plates. The concentration of compound was varied from 100 − 0.2 µg/ml. Incubation of plates was done for five days at 370C. 25 µl of 1:1 mixture of Alamar blue reagent and 10% Tween 80 was added after five days. Further incubation of 24 h was done. A blue color in well indicated the bacterial cell inhibition whereas pink color indicated cell growth. [xxx]
Molecular docking studies:
The structures of proteins used in this work were downloaded from the Protein Data Bank (PBD Code: 4Y6U). [] Molecular docking study has been carried out by the free software package Auto-dock Vina. [,] Initially, the protein structure was re-processed before using as receptor for docking. Necessary steps taken in preprocessing of protein were addition of hydrogen atoms, assignment of atomic charges, and elimination of water molecules that are not involved in ligand binding. The Auto-dock tool package was used for grid generation with a maximal size of 2700 Å with 0.6 Å spacing. Pymol was used to visualize the result of docking studies. []
{kind=link}
{kind=link}