Clinical presentations
Patient 1(P1, F-1 in Fig. 1A) is a female who was hospitalized at the age of 1-year-9-month-old due to experiencing fever and seizures three times in a single day. Upon physical examination, she exhibited developmental delay and left ankle clonus. Laboratory tests revealed elevated levels of lactate in the blood (16mM). Brain MRI indicated abnormalities in the bilateral dorsal thalamus, cerebellar dentate nucleus, and superior cerebellar peduncle. Echocardiography (ECG) revealed left ventricle enlargement and left ventricular wall thickening, which indicates hypertrophic cardiomyopathy. Genetic analysis revealed a GTPBP3 compound heterozygous mutation: c.689A > C inherited from the father and c.424G > A inherited from the mother.
Patient 2 (P2, F-2 in Fig. 1A) is a female born through full-term natural delivery. She exhibited delayed motor milestones and could not stand on her own at 9 months of age. At 1-year-1-month old, she was hospitalized due to vomiting for 4 consecutive days. Physical examination revealed hypotonia and hyperreflexia in the knee tendons. Blood lactate levels were elevated to 8.58mM. Brain MRI showed abnormal signals in the bilateral thalamus and peduncle. Electroencephalogram (EEG) indicated diffuse delta waves as the predominant slow wave pattern, and echocardiography revealed decreased left ventricular function (ejection fraction (EF) = 54%) and mild regurgitation of the second, tricuspid, and pulmonary valves. Genetic testing confirmed compound heterozygous variants in GTPBP3: c.934_957del inherited from her father and c.689A > C inherited from her mother.
Patient 3 (P3, F-3 in Fig. 1A) experienced a seizure with coma lasting 2 hours at the age of 3 years and 8 months. Her condition rapidly deteriorated, presenting with unconsciousness, abnormal breathing patterns, decreased oxygen saturation, and impaired liver function accompanied by uncorrectable metabolic acidosis. Laboratory examination revealed a blood glucose level of 27 mM, blood lactate level of 16mM, blood ammonia level of 94µM, alanine aminotransferase of 283.3U/L, aspartate aminotransferase of 716.6 U/L. Brain MRI revealed abnormal signals in the bilateral thalamus, peduncle, and bilateral temporoparietal cortex. EEG showed a slowing of basic waves, and ECG revealed that the left atrium and left ventricle were slightly enlarged, and the left ventricular systolic function was normal (EF = 55%, FS = 28%). Genetic examination revealed compound heterozygous variants in GTPBP3: c.689A > C (paternally inherited) and c.127C > T (maternally inherited).
Patient 4 (P4, F-4 in Fig. 1A) is a young boy who was admitted to the hospital at the age of 4 years and 8 months due to weakness that had persisted for over 2 years. He experienced fatigue easily, had poor endurance. Upon physical examination, it was noted that his growth and development were borderline normal, and he had astigmatism. Laboratory tests revealed a high-sensitive troponin-I level of 0.026 ng/mL, NT-proBNP of 894pg/ml, CK-MB of 21 U/L, and blood lactate level of 9.18mM. Ultrasonography showed left ventricular enlargement, ventricular wall hypertrophy, and an EF of 38%, consistent with a diagnosis of heart failure with grade III cardiac function. Genetic testing revealed a homozygous variant in GTPBP3 (c.473T > G).
Patient 5 (P5, F-5 in Fig. 1A) is a male infant who presented with rapid, shallow breathing and intermittent moaning starting at 20 hours after birth. His blood lactate level was significantly elevated at 26mM, indicating metabolic acidosis along with respiratory acidosis. Additionally, neonatal disease screening using LC/MS revealed a marked increase in alanine. Genetic examination revealed GTPBP3 compound heterozygous mutation: c.413C > T inherited from her father and c.509_510del inherited from her mother.
Patient 6 (P6, F-6 in Fig. 1A) is a female child born to healthy, unrelated parents. Her sibling passed away at 7 months due to a “brain disease”. Shortly after birth, developmental delays were noted in the child, presenting with symptoms of unsteady running, speech disorder, and limited comprehension. By the age of 2, she experienced sporadic seizures associated with colds and fever once or twice a year. Physical examination revealed increased muscle tone in the limbs, left eye esotropia, and restricted external rotation. Elevated blood lactic acid levels at 6.94 mmol/L were observed, along with abnormal signals in the bilateral thalamus and left cortex on brain MRI. Additionally, the ECG displayed two generalized spikes and slow spikes during sleep. Cardiac ultrasound indicated mild regurgitation in the tricuspid valve, main arteries, and pulmonary arteries. Genetic testing identified a compound heterozygous mutation in GTPBP3: c.187C > T (paternally inherited) and c.776A > G (maternally inherited).
Patient 7 (P7, F-7 in Fig. 1A) is a 3-year-old girl who was hospitalized due to multiple seizures. Genetic examination revealed GTPBP3 compound heterozygous mutation c.848C > A (paternally inherited) and c.680_691dup (maternally inherited).
Patient 8 (P8, F-8 in Fig. 1A), a male, was hospitalized at the age of 2 due to fever and seizures. He exhibited delayed motor development, weak muscle tone, hypotonia, and fatigue. Elevated blood lactate levels were recorded at 8.3 mM. Brain MRI displayed abnormal hyperintensity signals in the basal ganglia. Genetic examination revealed GTPBP3 compound heterozygous mutation c.689A > C (paternally inherited) and c.774_775insC (maternally inherited).
Patient 9 (P9, F-9 in Fig. 1A) is the third child born to unrelated parents. His sister is normal, and his brother passed away at the age of 1 year and two months of unknown etiology. He presented with language development delay and hypotonia of both lower limbs. Brain MRI revealed suspicious white matter abnormality. Genetic examination revealed GTPBP3 compound heterozygous mutation: c.689A > C (paternally inherited) and c.1092_1103del (maternally inherited).
Detailed clinical presentations and other examination results are summarized in Table 1.
Table 1
Clinical features of 9 patients with GTPBP3 deficiency. *LS, Leigh syndrome; MD, mitochondrial disease; NA, not available.
| Patient | P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | P9 |
| Gender | female | female | female | male | male | female | female | male | male |
| Age of onset | 1.8 years | 1.1 years | 3.7 years | 4.7 years | At bitrh (20h) | 7 months | 3 years | 2 years | 1 years |
| Variant | c.689A > C; c.424G > A | c.689A > C; c.934_957del | c.689A > C; c.127C > T | c.473T > G, hom | c.413C > T; c.509_510del | c.187C > T; c.776A > G | c.680_691dup; c.848C > A | c.774_775insC; c.689A > C | c.689A > C; c.1092_1103del |
| Clinical diagnosis | LS | LS | LS | MD | MD | LS | LS | MD | MD |
| Clinical features | developmental delay; seizure; fatigue | developmental delay; hypotonia; knee tendon hyperreflexes | Seizure; fatigue | developmental critical state; fatigue; exercise intolerance | rapid shallow breathing followed by intermittent moaning | developmental delay; epilepsy; hypertonia; knee tendon hyperreflexes | seizure | developmental delay; seizure; fatigue | developmental delay; hypotonia |
| Echocardiography (EF ≧ 50%;25%≦FS ≦ 45%) | cardiac hypertrophy (EF = 72%; FS = 40%) | low left ventricular function (EF = 54%; FS = 27%) | slightly larger atria and ventricles, low left ventricular function (EF = 55%; FS = 28%) | ventricular hypertrophy, slightly reduced left ventricular systolic function, mitral regurgitation (EF = 38–60%; FS = 28.5–45%) | NA | mild regurgitation of tricuspid valve and main and pulmonary valve | NA | NA | NA |
| Brain MRI | Bilateral thalamus, cerebellar dentate nucleus and local subcortical brain abnormal signals | Abnormal signals in the bilateral thalamus and peduncle | Bilateral high signal in thalamus, midbrain and peduncle | No obvious change | NA | Multiple asymmetric signal foci in the bilateral thalamus, brainstem and medulla oblongata | NA | Basal ganglia lesion | Suspected white matter abnormality |
| Plasma lactate level (mM) | 16 | 8.58 | 16 | 9.18 | NA | 5.92 | NA | 8.3 | NA |
| Others | NA | Abnormal electroencephalogram | Abnormal electroencephalogram | NA | NA | Abnormal electroencephalogram | NA | NA | NA |
Pathogenicity prediction of variants
Cross-species amino acid conservative analysis (Fig. 1B) was carried out among different variants. Except for c.689A > C (p.Q230P), other residues are highly conserved during evolution. The summary of pathogenicity analysis for genetic variants is presented in Table 2. gnomAD (http://gnomad.broadinstitute.org) was an allele frequency annotations database18. The variants were either absent or the frequency was extremely low in the population. SIFT (http://sift-dna.org) and PolyPhen-2 (http://genetics.bwh.harvard.eduy/pph2) were the typical pathogenicity prediction tools for non-synonymous single nucleotide substitution19,20. MutationTaster (https://www.mutationtaster.org) works on the DNA level, also suitable for indels. The score of c.413C > T, c.424G > A, c.473T > G, c.776A > G, and c.848C > A in SIFT all greater than 0.05. And the score of c.413C > T, c.424G > A, c.473T > G, c.776A > G and c.848C > A in PolyPhen-2 all greater than 0.909. Almost variants are predicted to be disease-causing in MutationTaster. Moreover, MUpro (http://mupro.proteomics.ics.uci.edu/) was used to predict the change in protein stability21. It seems that c.413C > T, c.424G > A, c.473T > G, c.689A > C, c.776A > G, and c.848C > A were more likely to lead to decreased protein stability.
Table 2
The annotations of variants.
| Variant (NM_032620.4) | Exon | Amino acid change | gnomAD | SIFT | Polyphen-2 | MutationTaster | MUpro |
| c.127C > T | 2 | p.Q43* | NA | NA | NA | D | NA |
| c.187C > T | 2 | p.R63* | NA | NA | NA | D | NA |
| c.413C > T | 4 | p.A138V | < 1‰ | 0.01 | 1.000 | D | decreased |
| c.424G > A | 4 | p.E142K | NA | 0.00 | 1.000 | D | decreased |
| c.473T > G | 4 | p.V158G | NA | 0.05 | 0.999 | D | decreased |
| c.509_510del | 4 | p.E170Gfs*42 | < 1‰ | NA | NA | D | NA |
| c.680_691dup | 6 | p.Q230_V231insGALQ | NA | NA | NA | P | NA |
| c.689A > C | 6 | p.Q230P | < 1‰ | 0.25 | 0.091 | D | decreased |
| c.774_775insC | 6 | p.N259Qfs*28 | < 1‰ | NA | NA | D | NA |
| c.776A > G | 6 | p.N259S | < 1‰ | 0.00 | 1.000 | D | decreased |
| c.848C > A | 7 | p.T283N | NA | 0.00 | 0.999 | D | decreased |
| c.934_957del | 7 | p.G312_V319del | < 1‰ | NA | NA | D | NA |
| c.1092_1103del | 8 | p.D364_R368del | NA | NA | NA | D | NA |
| *SIFT score: 0.0-0.05 means deleterious and 0.05-1.0 means tolerated; PolyPhen-2 score: 0.0-0.446 is Benign, 0.447–0.908 is possibly damaging and 0.909-1.0 for probably damaging; D-prediction disease causing, P-prediction polymorphism; NA: not found. |
In summary, a total of thirteen variant sites were involved in nine pedigrees, with five have been reported and eight novel variants. While predictive analyses indicate potential defects in all variants, confirmation through additional biological functional verification is required.
Decreased GTPBP protein levels and impaired mitochondrial function were observed in patient-derived immortalized lymphocytes
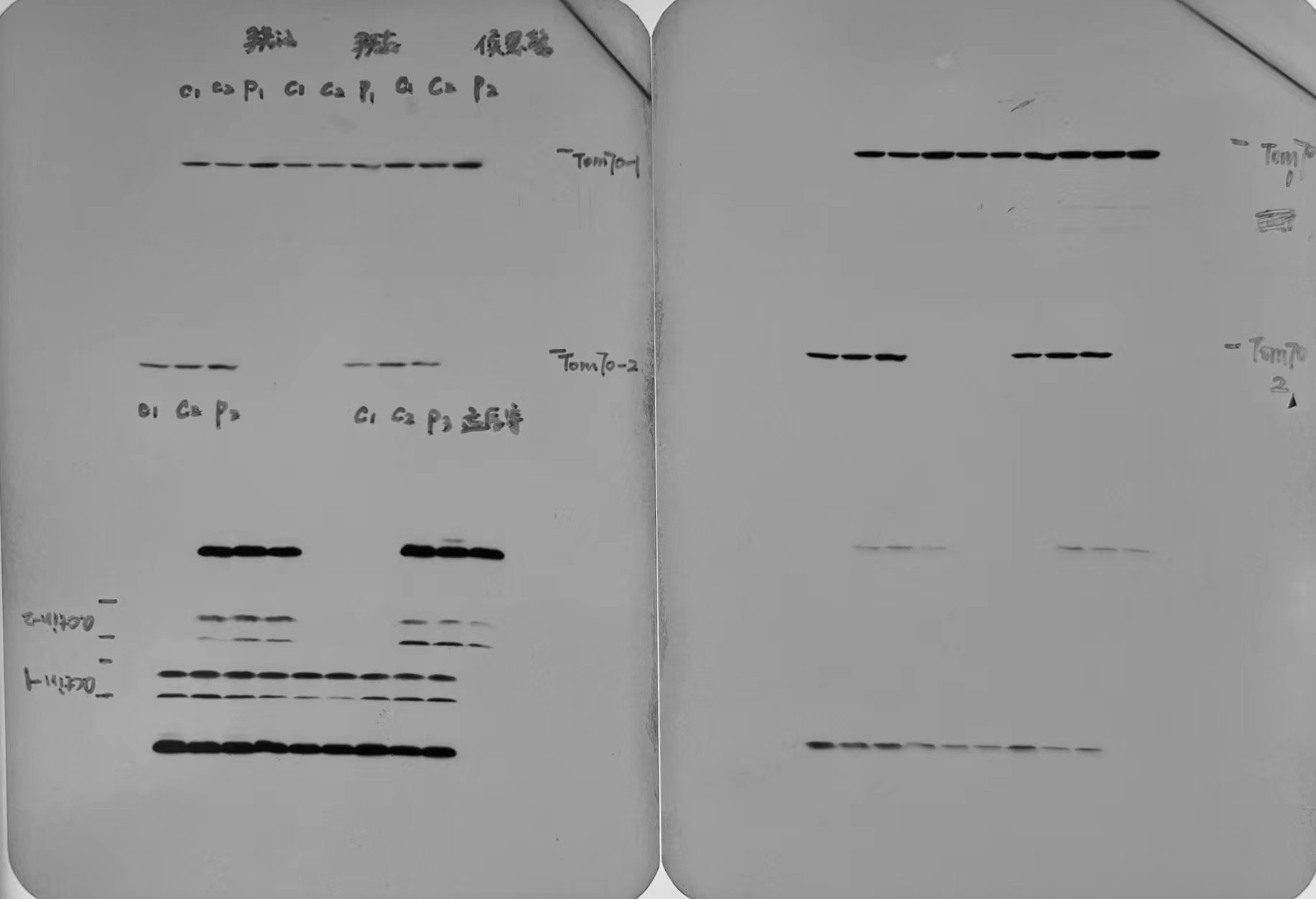
Four patients (P1-P4) and three age-matched healthy children as controls were included in the immortalized lymphocyte experiments. Initial validation of the immortalized lymphocytes was conducted through Sanger sequencing, confirming consistency with the previous genetic examination (Supplementary Fig. 1A). Analysis comparing the steady-state GTPBP3 protein levels in patients P1-P4 with the normal control group revealed significant decreases of 82.8% (p < 0.001), 79.1% (p < 0.001), 74.4% (p < 0.001), and 25.5% (p = 0.036) respectively (Fig. 2A-B). These findings highlight the substantial reduction in GTPBP3 protein levels in patient-derived lymphocytes.
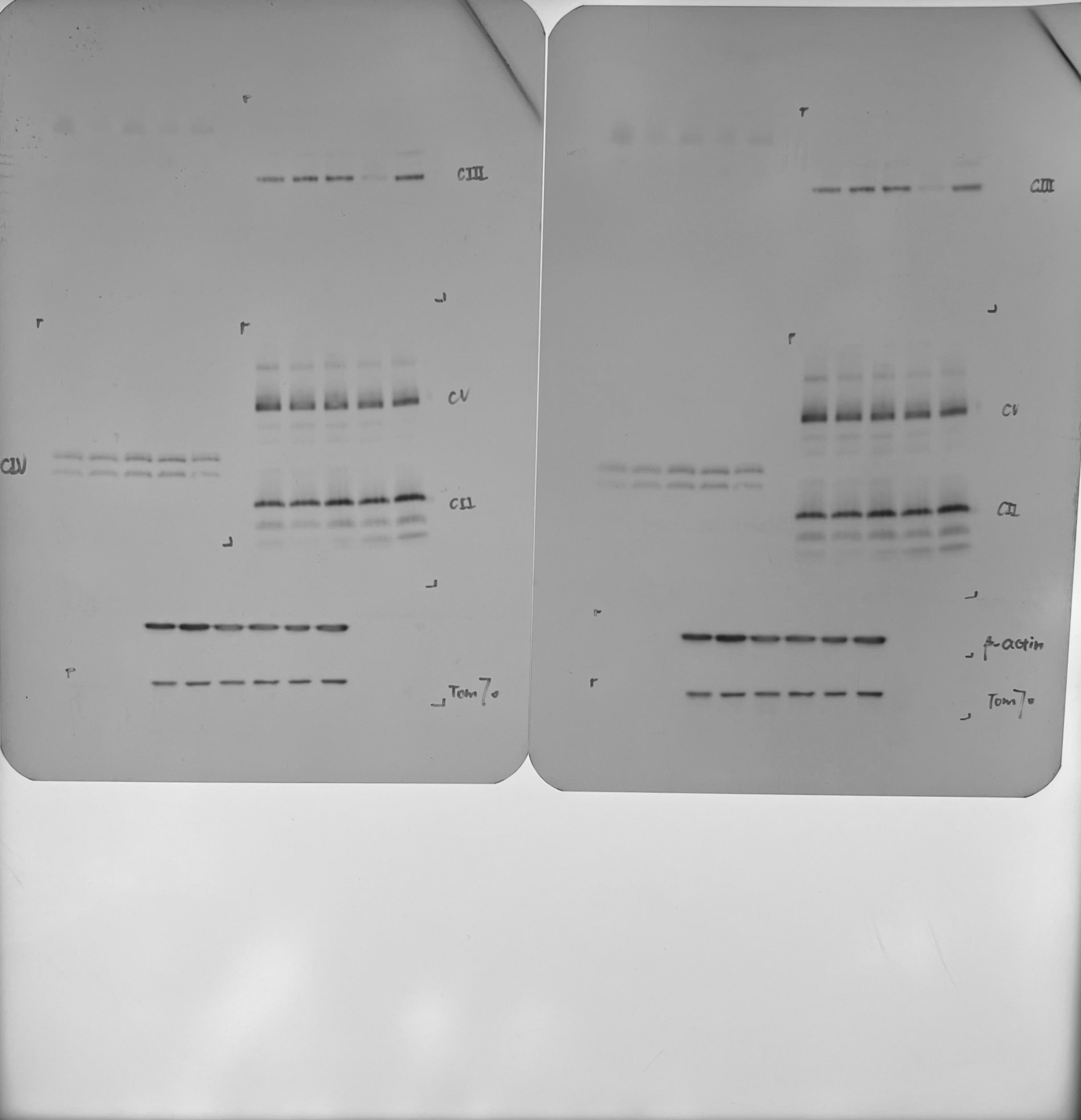
As previously mentioned, GTPBP3 is a highly conserved mt-tRNA modifying enzyme essential for the mitochondrial protein translation process7. To further elucidate its impact on mitochondrial function, BN-PAGE is a common technique optimized for the analysis of the five complexes (CI-CV) of OXPHOS (Fig. 2C-F)22,23. Compared with the control, the content of CI, CIII, CIV, and CV of P1 was decreased in P1 and P2. Additionally, in P4, the content of complex CIII was decreased, while no significant differences were observed in the abundance of mitochondrial complexes in P3. To further detect the mitochondrial respiratory capacity, the oxygen consumption levels in lymphocytes were measured24,25. The basal respiration rate was measured under normal conditions. Oligomycin was used to inhibit ATP synthase, allowing for the calculation of the corresponding OXPHOS-related oxygen consumption rate (OCR). FCCP was used to disrupt the proton gradient and mitochondrial membrane potential, stimulating cells to reach their maximum respiration potential26. As a result, basal OCR of P1was decreased by 23.7% (p < 0.001), P2 decreased by 30.2% (p < 0.001), P3 decreased by 20.4% (p < 0.001) and P4 decreased by 14.4% (p = 0.0012). The OCR of oxidative phosphorylation decreased by 57.8% (p < 0.001) in P1, 54.0% (p < 0.001) in P2, 47.4% (p < 0.001) in P3, and 57.4% (p < 0.001) in P4. The maximum respiration potential of P1 was decreased by 31.3% (p < 0.001), P2 was decreased by 29.9% (p < 0.001), P3 was decreased by 20.1% (p = 0.0018) and P4 was decreased by 19.4% (p = 0.0024) (Fig. 2E). In summary, the OXPHOS function of P1-P4 was impaired to varying degrees.
Re-expression of wild-type vectors rescues the deficit in GTPBP3 expression level and OXPHOS complexes
To further investigate the impact of GTPBP3 on mitochondrial functions, we utilized CRISPR-Cas9 technology to generate a HEK-293T GTPBP3 knockout (KO) cell model, which was then rescued by re-expressing wild-type GTPBP3. Western blot analysis confirmed the reduced level of GTPBP3 protein in the KO cell model (Fig. 3A). Additionally, blue native polyacrylamide gel electrophoresis (BN-PAGE) revealed significant decreases in Complexes I, III, IV, and V (Fig. 3B), consistent with findings in patient-derived lymphocyte models. Subsequent analysis of the re-expressed cells through WB and BN-PAGE demonstrated partial recovery of the observed defects (Fig. 3C-D).
Protein abundance decreased in GTPBP3 site-directed mutagenesis cell model
According to instructions of the Standard and guidelines for the interpretation of sequence (2015) published by the American Society for Medical Genetics and Genomics (ACMG)27, nonsense mutations, frameshifts, ± 1 or 2 canonical splice sites, initiation codons, and large deletions, all have pathogenic very strong evidence, combined with extremely low population frequency, they can be distributed to Likely pathogenic (LP) at least. Cytological function experiments can provide strong evidence for pathogenicity analysis, which was a milestone significance for VUS variants28,29. Re-expressing GTPBP3 carrying mutant vectors of c.127C > T, c.187C > T, c.473T > G, c.776A > G, c.848C > A and mutation hot spot (c.689A > C) vectors in GTPBP3 KO cells, mutations were identified by Sanger sequencing (Supplementary Fig. 1B).
To eliminate the interference of wild-type GTPBP3 protein, the GTPBP3 vectors carrying different mutations were transfected into KO cell lines. As shown in Fig. 4A, when compared with KO + GTPBP3 cell line, KO + c.127C > T decreased by 97.8% (p < 0.001), KO + c.187C > T decreased by 99.6% (p < 0.001), KO + c.424G > A decreased by 94.8% (p < 0.001), KO + c.473T > G decreased by 32.9% (p = 0.0064) and decreased by 45.7% (p < 0.001) in KO + c.689A > C. No significant decrease was found in KO + c.776A > G and KO + c.848C > A. It needs to be considered that there are differences in vector copy number among cell lines30,31. We designed primers targeting to the 3' end of the CDS and PGK promoter which was a conserved region on vectors to assess the relative level of vectors. As shown in Fig. 4B. The vector levels of each cell lines were compared with KO + GTPBP3 cell line, KO + c.127C > T was 3.2 times (p < 0.001), KO + c.187C > T was 2.4 times (p < 0.001), KO + c.424G > A was 1.8 times (p = 0.0118), KO + c.473T > G was 3.7 times (p < 0.001), and KO + c.689A > C was 2.8 times (p < 0.001), KO + c.776A > G was 2.5 times (p < 0.001), KO + c.848C > A was 3.2 times (p < 0.001), KO + c.1384C > G was 4.8 times (p < 0.001). Once the relative efficiency level further corrected the protein content, the masked differences of KO + c.776A > G and KO + c.848C > A can be uncovered (Fig. 4C).
Analysis of genetic variants spectrum and phenotype-genotype correlation of GTPBP3
The initial report by Robert Kopajtich et al in 2014 documented 11 cases of GTPBP3 mutations11. To date, in total of 21 cases with 23 distinct variants of GTPBP3 have been reported. By incorporating these reported variants with the 8 novel variants identified in our study, the genetic spectrum of the GTPBP3 gene was expanded (Fig. 5A). Notably, variants identified in the Chinese population are marked in yellow (Fig. 5A), we noted that c.689A > G is most common in the Chinese population. Moreover, our analysis revealed that the variant sites are predominantly concentrated in exon 4 and exon 6, with c.689A > C showing high frequencies of 8/51, indicating it as a hot spot mutation site within this population.
Based on the protein’s functional domains, the mutations can be roughly categorized into four regions, which are mitochondrial targeting sequence (MTS, M), GTP-box (G), C-terminal (C), and other undetermined (U) regions11. Combined with previous cases, the characteristics were summarized as follows (Fig. 5B-D). Firstly, the onset age for all cases was under 10 years old, with a trend to earlier onset in the M/U region (Fig. 5B). Secondly, the clinical outcomes of patients in the M/U region more servere, with a higher proportion mortality ratio (Fig. 5C). In terms of muscle involvement, individuals in the M/G/U region were usually affected by dual involvement. Those in the C region tended to exhibit muscle involvement, along with energy deficiency along with energy deficiency symptoms like fatigue and mild myocardial hypertrophy (limited by the small number of case samples) (Fig. 5D). Overall, it appears that symptoms in the C region were relatively mild, may due to the smaller sample size. Lastly, all patients exhibited hyperlactatemia, and the majority experienced developmental delay along with muscle and/or nerve involvement (such as cardiac hypertrophy, seizures, hypermyotonia, or hypotonia) and abnormal brain MRI. They may also present with dyspnea, feeding difficulties, short stature, and occasionally visual impairment, as well as cardiac abnormalities. These cardiac abnormalities can include conduction and heart valve issues. Notably, valvular insufficiency was initially linked to GTPBP3 deficiency in our study.
{kind=link}
{kind=link}