3.1 Optimized Geometry
The optimized configuration of compound 1 and 2 is of paramount significance as it unveils crucial insights into their molecular arrangement and spatial conformation. The geometry not only provides a visual representation of the structural features (Fig. 2) but also offers valuable information about intermolecular interactions and overall three-dimensional characteristics (Shinggu et al., 2023b). Optimization helps in understanding the selected compounds' properties, including stability, reactivity, and binding interactions (Venkatesh et al., 2000; Onen et al., 2017). In essence, the optimized geometry serves as a key to unraveling the functional aspects of compound 1 and 2, contributing to a comprehensive comprehension of their molecular nature and potential applications.
Table 1
Optimized Geometry of Compounds 1, & 2 using Solvents of DMSO, Ethanol and Methanol
| Bond Lengths | Compound 1 | Bond Lengths | Compound 2 |
| DMSO | Ethanol | Methanol | DMSO | Ethanol | Methanol |
| R(1–8) | 1.236 | 1.236 | 1.240 | R(1–10) | 1.367 | 1.367 | 1.367 |
| R(2–9) | 1.237 | 1.237 | 1.242 | R(2–13) | 1.359 | 1.359 | 1.362 |
| R(3–8) | 1.409 | 1.409 | 1.403 | R(3–7) | 1.426 | 1.425 | 1.432 |
| R(3–9) | 1.408 | 1.407 | 1.403 | R(3–21) | 0.974 | 0.974 | 0.973 |
| R(3–10) | 1.448 | 1.448 | 1.449 | R(4–12) | 1.363 | 1.363 | 1.363 |
| R(4–10) | 1.361 | 1.362 | 1.360 | R(4–25) | 0.966 | 0.966 | 0.968 |
| R(4–15) | 1.007 | 1.007 | 1.007 | R(5–14) | 1.359 | 1.359 | 1.361 |
| R(4–16) | 1.006 | 1.006 | 1.006 | R(5–26) | 0.968 | 0.968 | 0.969 |
| R(5–10) | 1.288 | 1.287 | 1.287 | R(6–9) | 1.464 | 1.464 | 1.465 |
| R(5–17) | 1.024 | 1.024 | 1.024 | R(6–15) | 1.466 | 1.466 | 1.466 |
| R(6–7) | 1.536 | 1.536 | 1.537 | R(6–20) | 1.016 | 1.016 | 1.017 |
| R(6–8) | 1.513 | 1.513 | 1.509 | R(7–8) | 1.513 | 1.513 | 1.512 |
| R(6–11) | 1.089 | 1.089 | 1.090 | R(7–9) | 1.540 | 1.541 | 1.536 |
| R(6–12) | 1.090 | 1.090 | 1.090 | R(7–16) | 1.098 | 1.098 | 1.096 |
| R(7–9) | 1.512 | 1.512 | 1.508 | R(8–10) | 1.388 | 1.388 | 1.389 |
| R(7–13) | 1.089 | 1.089 | 1.090 | R(8–11) | 1.398 | 1.398 | 1.399 |
| R(7–14) | 1.090 | 1.090 | 1.090 | R(9–17) | 1.094 | 1.094 | 1.094 |
| - | - | - | - | R(9–18) | 1.099 | 1.099 | 1.100 |
| - | - | - | - | R(10–12) | 1.390 | 1.390 | 1.392 |
| - | - | - | - | R(11–13) | 1.383 | 1.383 | 1.381 |
| - | - | - | - | R(11–19) | 1.081 | 1.081 | 1.082 |
| - | - | - | - | R(12–14) | 1.399 | 1.399 | 1.399 |
| - | - | - | - | R(13–14) | 1.392 | 1.392 | 1.391 |
| - | - | - | - | R(15–22) | 1.093 | 1.093 | 1.094 |
| - | - | - | - | R(15–23) | 1.100 | 1.100 | 1.101 |
| - | - | - | - | R(15–24) | 1.092 | 1.092 | 1.093 |
Understanding the bond lengths during the optimization of a compound in various solvation methods is a critical aspect of molecular characterization (Thamarai et al., 2020). The variations in bond lengths provide valuable insights into the dynamic interplay between the compound's molecular structure and the surrounding solvent environment. These changes reflect the compound's adaptability to different solvents, shedding light on its stability, reactivity, and potential interactions within diverse chemical surroundings (Adindu et al., 2023).
The presented table (Table 1) outlines bond lengths in Compound 1 and 2 across three solvents - Dimethyl Sulfoxide (DMSO), Ethanol, and Methanol. Optimizing these compounds in Dimethyl Sulfoxide (DMSO), Ethanol, and Methanol solvents is a crucial process aimed at comprehending and defining their behavior in diverse environments. Both compound 1 & 2 bonds display slight variations in length, with Dimethyl Sulfoxide (DMSO) and Ethanol yielding similar values, while methanol results in a slightly longer bond. Certain bonds like R(7–9) and among other bonds in compound 1 and 2 have same bond length with Dimethyl Sulfoxide (DMSO) and ethanol but slight variation in methanol. This observation implies that the nature of the solvent can influence the specific interatomic distances within the molecules, with Dimethyl Sulfoxide (DMSO) and ethanol showing a similar impact on bond lengths, while methanol induces a subtle but discernible alteration which further suggest that a notable insensitivity of both compounds to variations in the solvent, particularly within the tested contexts of Dimethyl Sulfoxide (DMSO), ethanol and methanol as methanol only shows a non-significant variations. The molecule's properties, encompassing factors like geometry and other molecular descriptors, demonstrate a remarkable uniformity across these solvents. This coherence in results implies that both compounds likely possesses a stable molecular structure, exhibiting comparable interactions and conformations in both Dimethyl Sulfoxide (DMSO), ethanol and methanol.
3.2 Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) Energy Levels
Table 2
Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) energy levels of the Compounds in Dimethyl Sulfoxide (DMSO), Ethanol and Methanol Solvent. Highest Occupied Molecular Orbital and Lowest Unoccupied Molecular Orbital (HOMO-LUMO) underscores the significance of solvation conditions in understanding the molecular properties and behavior of these compounds, offering valuable insights for applications in diverse chemical environments (Odey et al., 2023).
| Molecule | Solvation Method | HOMO Energy(eV) | LUMO Energy(eV) |
| Compound 1 | DMSO | -6.7882 | -0.9072 |
| Methanol | -6.8883 | -1.2694 |
| Ethanol | -6.8037 | -0.9355 |
| Compound 2 | DMSO | -6.3362 | -0.4373 |
| Methanol | -6.4807 | -0.5818 |
| Ethanol | -6.3362 | -0.4373 |
Table 2 outlines the HOMO and LUMO energy levels for Compound 1 and Compound 2 in different solvation methods; Dimethyl Sulfoxide (DMSO), Methanol, and Ethanol, obtained through quantum chemical calculations and measured in in electron volts (eV).
Comparing HOMO energy across solvents reveals a slight variation, with the most negative value in Methanol (-6.8883 eV), followed by Ethanol (-6.8037 eV) and Dimethyl Sulfoxide (DMSO) (-6.7882 eV). More negative HOMO energies indicate a greater tendency to donate electrons, suggesting that compound 1 is most electron-donating in Methanol, followed by Ethanol and DMSO. Similarly, LUMO energy follows a comparable trend, with the most negative value in Methanol (-1.2694 eV), followed by Ethanol (-0.9355 eV), and DMSO (-0.9072 eV). Lower LUMO energies signify a higher electron-accepting ability, indicating that compound 1 with methanol solvation is the most electron-accepting, followed by Ethanol and Dimethyl Sulfoxide (DMSO). These variations across solvents highlight the impact of the solvation environment on compound 1’s electronic structure and reactivity, crucial information for understanding its behavior in different settings and guiding the optimization of processes involving the compound.
In a similar vein, when considering Compound 2, an analysis of HOMO energy levels indicates minimal fluctuation across solvents. The HOMO energy remains consistently steady in both Dimethyl Sulfoxide (DMSO) (-6.3362 eV) and Ethanol (-6.3362 eV), with a slightly more negative value in Methanol (-6.4807 eV). This suggests that the molecule's inclination to donate electrons is not significantly altered by the solvent choice. Similarly, the LUMO energy levels exhibit subtle variations, with the most negative value observed in Methanol (-0.5818 eV), followed by Dimethyl Sulfoxide (DMSO) (-0.4373 eV) and Ethanol (-0.4373 eV). These nuanced changes imply that the molecule's capacity to accept electrons is only subtly affected by the solvent. The consistent HOMO and LUMO energy levels suggest that the electronic structure and reactivity of compound 2 relatively stable across the solvents studied. While these solvent-induced alterations are modest compared to those in other molecules, they still bear significance for the compound's behavior in diverse chemical environments. A comprehensive understanding of the electronic characteristics in different solvents is imperative for predicting and managing the molecule's reactivity in various applications or reaction conditions.
In Figs. 3 and Fig. 4, the HOMO-LUMO diagrams of Compound 1 and Compound 2 are presented, offering visual representations of their respective electronic structures.
3.3 Vibrational Frequencies
Table 3
Vibrational Frequencies of Compound 1
| Vibrational Mode | Frequency (cm) |
| DMSO Solvent | Ethanol Solvent | Methanol Solvent |
| C-N | 1001.57 | 93.4343 | 39.5346 |
| C = O | 870.10 | 102.1166 | 94.7053 |
| 1145.16 | 108.5369 | 107.5694 |
| 1210.31 | 188.4963 | - |
| C-H | 434.58 | 311.543 | 421.3864 |
| 559.04 | 431.007 | 459.7723 |
| 6721.51 | 475.3764 | 468.6184 |
| 688.19 | 499.2952 | 497.0187 |
| 730.77 | 729.009 | - |
| C-C | 1475.99 | 553.7869 | 551.7419 |
| C = C | 553.04 | 559.8785 | 559.237 |
| 600.11 | 599.76 | 603.7468 |
| 1066.14 | 643.18 | 1066.903 |
3.3.1 The Vibrational Spectrum of Compound 1 In DMSO Solvent
The vibrational spectrum of compound 1 (Fig. 5), the frequencies and intensities provide a comprehensive understanding of the compound's structural composition and its interaction with the solvent, methanol. Each peak corresponds to specific functional groups, and the variations in intensity highlight the compound's sensitivity to the solvent environment. In the vibrational spectrum (Table 3) of compound 1 in Dimethyl Sulfoxide (DMSO) solvent, several prominent peaks provide valuable insights into its molecular composition. The peak at 1001.57 cm⁻¹ signifies a strong C-N stretch vibration, characteristic of the carbon-nitrogen bond, suggesting the presence of a guanidine group. Guanidine compounds typically exhibit robust C-N stretching vibrations, making this peak indicative of the specific nitrogen-containing functional group within the molecule. Moving on to carbon-oxygen vibrations, two distinct peaks stand out. The first, at 870.10 cm⁻¹, corresponds to a potent C = O stretch vibration commonly associated with carbonyl groups. This suggests the presence of a carbonyl group, potentially in the moiety of compound 1. The second peak at 1145.16 cm⁻¹ hints at another C = O stretch vibration, possibly associated with a different carbonyl group within the compound. Additionally, the peak at 1210.31 cm⁻¹ suggests a third C = O stretch vibration, highlighting the structural complexity of the molecule. The carbon-hydrogen (C-H) vibrations in the range of 434.58 cm⁻¹ to 730.77 cm⁻¹ unveil the intricate hydrocarbon framework of compound 1. Peaks at 434.58 cm⁻¹, 559.04 cm⁻¹, 672.51 cm⁻¹, 688.19 cm⁻¹, and 730.77 cm⁻¹ indicate diverse C-H bend or stretch vibrations, reflecting the presence of various types of hydrogen atoms within the molecule. This multiplicity underscores the complexity of the hydrocarbon moieties contributing to the overall vibrational spectrum. The C-C stretch vibration at 1475.99 cm⁻¹ suggests the existence of carbon-carbon bonds, potentially within the carbon backbone or cyclic structures of the molecule. Lastly, the C = C vibrations at 553.04 cm⁻¹, 600.11 cm⁻¹, and 1066.14 cm⁻¹ indicate the presence of carbon-carbon double bonds, contributing to the overall molecular geometry of compound 1. These double bonds play a crucial role in the compound's reactivity and chemical behavior. The vibrational spectrum of compound 1 unveils a rich array of functional groups, including the distinctive guanidine group, carbonyl moieties, diverse hydrocarbon structures, carbon-carbon bonds, and carbon-carbon double bonds. Each peak in the spectrum provides valuable information about the compound's structural composition and potential reactivity.
3.3.2 The Vibrational Spectrum of Compound 1 In Ethanol Solvent
The vibrational spectrum of compound (Table 3, Fig. 6) 1 optimized in ethanol, distinct shifts and intensity variations in certain peaks suggest a notable influence of the solvent on the molecular vibrations. Starting with the C-N (carbon-nitrogen) vibrations, the peak at 93.4343 cm⁻¹ indicates a C-N stretch vibration, characteristic of the carbon-nitrogen bond. The heightened intensity at this frequency suggests a pronounced interaction within the molecule, possibly influenced by the solvent, ethanol. The solvent-solute interactions in the C-N bond may play a significant role in altering the observed vibrational characteristics. Moving to the C = O (carbon-oxygen) vibrations, the peaks at 102.1166 cm⁻¹, 108.5369 cm⁻¹, and 188.4963 cm⁻¹ represent C = O stretch vibrations associated with carbonyl groups. The shifts and intensity variations in these peaks compared to the previous spectrum point to changes in the molecular environment due to the presence of ethanol. The solvent appears to induce modifications in the carbonyl environments, possibly influencing the number and nature of carbonyl groups within compound 1. The C-H (carbon-hydrogen) vibrations, spanning from 311.543 cm⁻¹ to 499.2952 cm⁻¹, exhibit consistent patterns, reflecting the diverse hydrocarbon moieties in the compound. However, variations in intensity, notably at 431.007 cm⁻¹ and 475.3764 cm⁻¹, indicate the sensitivity of C-H vibrations to the solvent environment. Ethanol, as a polar solvent, may interact differently with various hydrocarbon groups, leading to changes in the observed C-H vibrational characteristics. Examining the C-C (carbon-carbon) vibrations at 553.7869 cm⁻¹, the shift in position and intensity changes compared to the previous spectrum suggests alterations in the molecular structure influenced by the solvent. Ethanol interactions may impact the carbon-carbon bonds, influencing the overall conformation of compound 1. Lastly, the C = C (carbon-carbon double bond) vibrations at 559.8785 cm⁻¹, 599.76 cm⁻¹, and 643.18 cm⁻¹ indicate the presence of double bonds, and their positions and intensities are sensitive to solvent effects. The changes observed in these peaks suggest structural adjustments in the presence of ethanol, highlighting the dynamic nature of the compound-solvent interaction. In summary, the vibrational spectrum of compound 1 in ethanol provides valuable insights into the solvent-induced changes in molecular vibrations. Ethanol alters the positions and intensities of specific vibrational modes, revealing the compound's responsiveness to the solvent environment and offering crucial information about its behavior in different solvents.
3.3.3 The Vibrational Spectrum of Compound 1 In Methanol Solvent
In the vibrational spectrum (Table 3, Fig. 7) of compound 1 optimized in methanol as the solvent, we observe distinctive changes in the molecular vibrations compared to the previous solvent-optimized spectrum. Beginning with the C-N (carbon-nitrogen) vibrations at 39.5346 cm⁻¹, the potential C-N stretch vibration indicative of the carbon-nitrogen bond is still present. However, the low intensity suggests that other vibrations may contribute to this frequency, emphasizing the intricate nature of nitrogen-containing functional groups within the molecule. Moving to the C = O (carbon-oxygen) vibrations, the peaks at 94.7053 cm⁻¹ and 107.5694 cm⁻¹ signify C = O stretch vibrations associated with carbonyl groups. The moderate intensity at 94.7053 cm⁻¹ suggests the presence of this functional group, while the higher intensity at 107.5694 cm⁻¹ indicates a more significant contribution, possibly from a different carbonyl group within compound 1. The use of methanol as the solvent introduces a distinct molecular environment, influencing the observed vibrational modes of carbonyl groups. In the C-H (carbon-hydrogen) vibrations, the peaks at 316.7589 cm⁻¹, 421.3864 cm⁻¹, 459.7723 cm⁻¹, 468.6184 cm⁻¹, and 497.0187 cm⁻¹ indicate C-H bend or stretch vibrations. The higher intensity at 421.3864 cm⁻¹ emphasizes a significant contribution from C-H vibrations, highlighting the complex hydrocarbon framework in compound 1. Changes in intensity and position, especially at 459.7723 cm⁻¹ and 468.6184 cm⁻¹, suggest a sensitivity of C-H vibrations to the methanol solvent, influencing the molecular interactions within the hydrocarbon moieties. Examining the C-C (carbon-carbon) vibrations at 551.7449 cm⁻¹, there is a distinct shift in position and intensity compared to the previous solvent-optimized spectrum, indicating alterations in the molecular structure influenced by methanol. The solvent-induced changes in the C-C vibrations underscore the impact of the choice of solvent on the compound's conformation. Finally, focusing on the C = C (carbon-carbon double bond) vibrations at 559.237 cm⁻¹ and 603.7468 cm⁻¹, these peaks showcase the sensitivity of double bond vibrations to methanol effects. The shifts in position and intensity suggest structural adjustments influenced by the solvent, providing insights into the dynamic behavior of compound 1 in a methanol environment. In summary, the vibrational spectrum of compound 1 in methanol reveals notable shifts and intensity variations compared to the previous solvent-optimized spectrum. The choice of methanol as the solvent introduces unique interactions that influence the compound's molecular vibrations, providing valuable information about its behavior in diverse solvent environments.
Table 4
Vibrational Frequencies of Compound 2 in DMSO, Ethanol and Methanol Solvent.
| Vibrational Mode | Frequency (cm) |
| DMSO Solvent | Ethanol Solvent | Methanol Solvent |
| C-F | 237.617 | 95.5276 | 58.054 |
| C-O | 247.9894 | 248.3084 | 122.358 |
| 1488.7221 | 1488.8298 | 191.6524 |
| O-H | 359.208 | 290.8983 | 637.8308 |
| C-C | 156.2992 | 156.4713 | 718.2607 |
| C = C | 126.453 | 198.6966 | 977.6243 |
| N-H | 3505.05 | 3505.2783 | 3495.9037 |
3.3.4 The Vibrational Spectrum of Compound 2 In DMSO Solvent
The infrared spectrum of compound 2, recorded in Dimethyl Sulfoxide (DMSO) solvent and elucidates the compound's molecular composition through distinct vibrational modes (Table 4, Fig. 8). The most noteworthy peak at 237.617 cm⁻¹ signifies C-F stretching vibrations, underscoring the presence of robust fluorine-carbon bonds. With an intensity of 104.4944, this peak suggests a substantial contribution from these bonds, offering insights into the compound's structural characteristics. The second significant peak at 247.9894 cm⁻¹ corresponds to intense C-O stretching vibrations, pointing towards the involvement of carbonyl or hydroxyl groups within the molecular framework. The high intensity of 175.5993 emphasizes the prominence of oxygen-containing functional groups in the compound. Together, these first two peaks provide a strong foundation for understanding the compound's chemical structure, hinting at the presence of both fluorine and oxygen-related moieties. Moving forward, the third major peak at 156.2992 cm⁻¹ indicates significant C-C stretching vibrations, highlighting the presence of carbon-carbon bonds. With an intensity of 6.2459, this peak contributes notably to the overall spectrum, showcasing the importance of carbon-carbon interactions in the molecular architecture. Furthermore, additional peaks associated with C = C stretching, O-H stretching, and N-H stretching vibrations provide a comprehensive overview of the compound's functional groups and their relative abundances, deepening our understanding of its chemical nature. The infrared spectrum of compound 2 in ethanol solvent reveals a rich tapestry of vibrational modes associated with C-F, C-O, O-H, C-C, C = C, and N-H stretching vibrations. The intensity variations in these peaks offer valuable information about the prevalence of specific functional groups, providing a detailed molecular fingerprint that enhances our comprehension of the compound's structure and potential reactivity.
3.3.5 The Vibrational Spectrum of Compound 2 In Ethanol Solvent
Table 4 and Fig. 8 outlines the infrared (IR) spectrum of compound 2, analyzed in ethanol solvent, reveals a diverse array of vibrational modes associated with specific functional groups. The most prominent peak, with an intensity of 95.5276 cm⁻¹, corresponds to C-F stretching vibrations, suggesting the presence of strong fluorine-carbon bonds in the compound. Additionally, the second highest intensity peak at 248.3084 cm^-1 indicates significant C-O stretching vibrations, indicative of carbonyl or hydroxyl groups. The third major peak at 156.4713 cm⁻¹ corresponds to C-C stretching vibrations, revealing the presence of carbon-carbon bonds. Further exploration of the spectrum reveals several other key vibrational modes. Peaks at 198.6966 cm⁻¹ and 290.8983 cm⁻¹ are associated with C = C stretching and O-H stretching vibrations, respectively, suggesting the presence of double bonds and hydroxyl groups. The spectrum also exhibits peaks corresponding to N-H stretching and bending vibrations, C-H stretching and bending vibrations, and C-N stretching vibrations, pointing to the existence of amine groups, alkyl groups, and nitrogen-carbon bonds in the molecular structure. The IR spectrum of compound 2 in ethanol solvent provides valuable insights into its molecular composition. The intense peaks at specific frequencies indicate the abundance of distinct functional groups, such as fluorine-carbon bonds, carbonyl or hydroxyl groups, and carbon-carbon bonds. The presence of amine groups, double bonds, and alkyl groups further contributes to the complexity of the molecular structure, enhancing our understanding of the compound's chemical composition and potential reactivity.
3.3.6 The Vibrational Spectrum of Compound 2 In Methanol Solvent
Table 4 and Fig. 9 outlines the vibrational spectrum of compound 2, the frequencies and intensities provide a comprehensive understanding of the compound's structural composition and its interaction with the solvent, methanol. Each peak corresponds to specific functional groups, and the variations in intensity highlight the compound's sensitivity to the solvent environment. The peaks in the C-F (Carbon-Fluorine) vibrations, such as the one at 58.054 cm⁻¹, suggest potential C-F stretch vibrations, indicating the presence of carbon-fluorine bonds. The low intensity may imply a moderate abundance of this functional group, with the possibility of interactions influenced by the methanol solvent. In the C-O (Carbon-Oxygen) vibrations, peaks at 122.358 cm⁻¹ and 191.6524 cm⁻¹ represent C-O stretch vibrations associated with hydroxyl and other oxygen-containing groups, respectively. The moderate and strong intensities suggest significant contributions from these functional groups, and variations in intensity may be attributed to interactions with the methanol solvent. The O-H (Hydroxyl) vibration peak at 637.8308 cm⁻¹ indicates O-H stretch vibrations characteristic of hydroxyl groups. The high intensity implies a substantial presence of hydroxyl functional groups in the molecule, with potential variations influenced by the methanol solvent. The C-C (Carbon-Carbon) vibration peak at 718.2607 cm⁻¹ suggests a C-C stretch vibration, reflecting the presence of carbon-carbon bonds. The substantial intensity implies a significant contribution from C-C vibrations, and changes may be associated with the solvent environment. The C = C (Carbon-Carbon Double Bond) vibration peak at 977.6243 cm⁻¹ represents a C = C stretch vibration indicative of carbon-carbon double bonds. The high intensity implies a significant contribution from double bond vibrations, with potential adjustments influenced by the methanol solvent. The vibrational frequencies of compound 2 in methanol reveal a complex interplay between the compound's functional groups and the solvent. The intensity variations underscore the compound's sensitivity to the methanol environment, providing valuable insights into its structural dynamics and behavior in different solvent conditions.
3.3.7 Comparison of the Vibrational Spectrum of Compound 1 & 2
While comparing the vibrational properties of both compounds, Compound 2 exhibits unique vibrational modes not present in Compound 1. For example, in methanol solvent, Compound 2 shows a significant vibrational mode at 718.2607 cm⁻¹ corresponding to C-C stretching, which is absent in Compound 1. Conversely, Compound 1 displays unique vibrational modes not found in Compound 2. For example, in DMSO solvent, Compound 1 shows a vibrational mode at 6721.51 cm⁻¹, which is likely associated with C-H stretching and is not observed in Compound 2.
In the DMSO solvent, Compound 2 exhibits higher frequencies for most vibrational modes compared to Compound 1. For example, the C-F, C-O, and O-H frequencies are notably higher in Compound 2. In contrast, in ethanol and methanol solvents, Compound 1 generally shows higher frequencies for most vibrational modes compared to Compound 2. For instance, in ethanol solvent, the C-N and C = O frequencies are higher in Compound 1.
Despite differences in solvent dependence, some vibrational modes are similar between the two compounds. For instance, in ethanol solvent, both compounds exhibit similar frequencies for C = C stretching vibrations, albeit with slight variations. Overall, the comparison highlights how the choice of solvent can significantly influence the vibrational frequencies of compounds, and it underscores the importance of considering solvent effects in vibrational spectroscopy studies.
3.4 Molecular Docking Studies
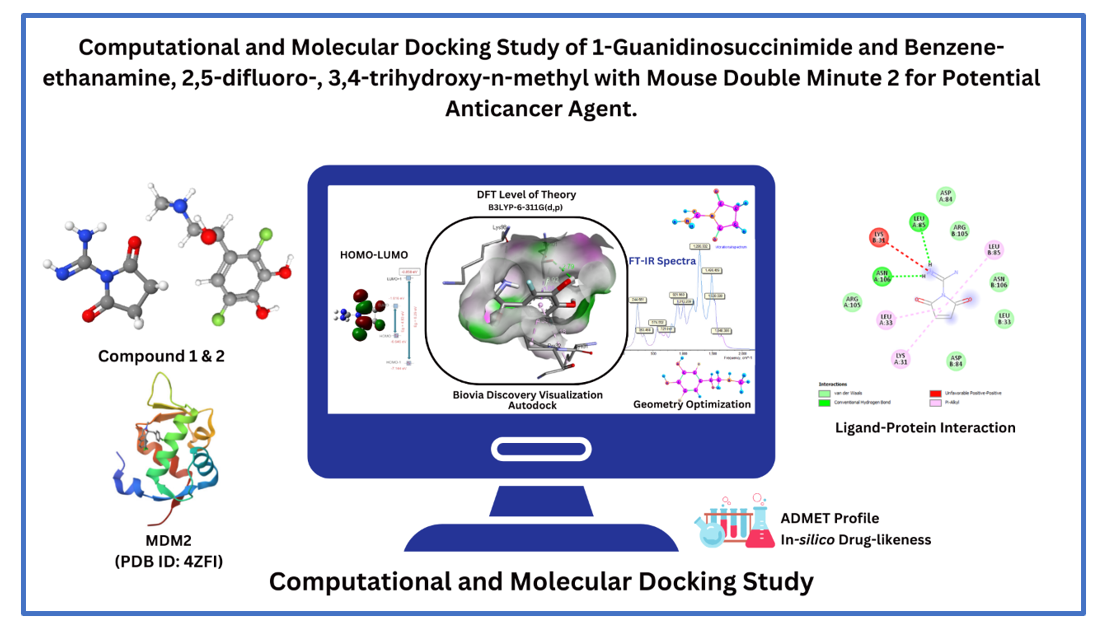
Molecular docking studies are generally utilized to explore binding energy and validate the molecular mechanisms of ligands at a protein's active site (Guedes et al., 2014). In this study, two distinct compounds (designated as compound 1 & 2) underwent molecular docking against a chosen protein, namely 4zfi, employing AutoDock Vina command prompt to elucidate the binding modes and the possible interactions.
Table 5
Molecular Docking Results of Compound 1, & 2 against Mouse Double Minute 2 (PDB ID: 4ZFI)
| Compounds | Protein Code | Binding Affinity (kcal/mol) | H bonds | Residue Interactions |
| Hydrophobic and Electrostatic Interactions | Van der Waal’s Interactions |
| Compound 1 | 4ZFI | -5.9 | Conventional-Hydrogen-A:LEU85 (2.36 Å), and Conventional-Hydrogen-A:ASN106 (2.14 Å) | Unfavorable positive-positive Pi-Alkyl-A:LYS31 (5.12 Å), Pi-Alkyl-A:LEU33 (5.22 Å), Pi-Alkyl-B:LEU85 (5.38 Å) | A:ASP 84, B:ARG 105, B:ASN 106, B:LEU 33, B:ASP 84, A:ARG 105. |
| Compound 2 | 4ZFI | -6.6 | Conventional-Hydrogen-D:THR101 (2.79 Å), Carbon-Hydrogen-D:LYS98 (3.34 Å), and Pi-Donor-Hydrogen-D:THR101 (3.92Å) | Hydrophobic-Pi-Sigma-D:THR101 (3.88 Å), Hydrophobic-Pi-Alkyl-B:LYS31 (5.18 Å), and Hydrophobic-Pi-Alkyl-B:PRO32 (4.38 Å) | - |
Keys
leusine (LEU), asparagine (ASN), lysine (LYS), aspartic acid (ASP), arginine (ARG), Threonine (THR), Proline (PRO).
Table. 5 reveals the results obtained from the molecular docking study of 1-guanidinosuccinimide (1-GSI) with the protein 4ZFI. The findings indicate that 1-GSI binds to 4ZFI with a binding affinity of -5.9 kcal/mol. This binding affinity is attributed to a combination of interactions, including hydrogen bonds, pi-alkyl interactions, hydrophobic interactions, and van der Waal's interactions.
Compound 1 (1-Guanidinosuccinimide) forms two conventional hydrogen bonds with residues LEU85 (2.36 Å) and ASN106 (2.14 Å) within the binding site of 4ZFI. These hydrogen bonds are anticipated to play a pivotal role in enhancing the stability of the complex, imparting specific and directional interactions crucial for ligand-protein binding (Morozov & Kortemme, 2005). This assertion gains support from the observation that 1-Guanidinosuccinimide is involved in pi-alkyl interactions with residues LYS31 (5.12 Å), albeit characterized as unfavorable positive-positive Pi-Alkyl interactions. Despite their unfavorable nature, these interactions may introduce distinctive nuances to the overall binding dynamics. Additionally, the ligand engages in pi-alkyl interactions with LEU33 (5.22 Å) and LEU85 (5.38 Å), where the pi-electrons of the ligand's aromatic ring interact with the alkyl side chains of the amino acid residues. These interactions significantly contribute to the overall binding affinity, providing non-polar interactions that effectively stabilize the ligand-protein complex.
In addition to the aforementioned interactions, 1-Guanidinosuccinimide also participates in van der Waal's interactions with residues ASP84, ARG105, ASN106, and LEU33. These interactions are weak but additive, meaning that the sum of all van der Waal's interactions contributes to the overall binding affinity. These interactions provide close-range attractions between the ligand and the protein, further stabilizing the complex.
The 2D and 3D ligand-protein interactions of compound 1 with 4ZFI are illustrated in Fig. 11. The 3D ball and stack models delineate the binding pocket structure of 4ZFI in association with compound 1. Hydrogen bonds formed between the compound and amino acids are represented by green dashed lines, an unfavoarable positive-positive interactions depicted with a red dashed lines, while pi-alkly interactions are depicted by pink dashed lines purple and van der Waals intereactions are deepicted as light green.
Compound 2 (Benzene-ethanamine, 2,5-difluoro-, 3,4-trihydroxy-n-methyl) exhibits a binding affinity of -6.6 kcal/mol towards protein 4ZFI as depicted in Table 5, implying a tight and stable interaction between the compound and the protein. This interaction is further reinforced by the presence of three hydrogen bonds: a conventional hydrogen bond with THR101 (2.79 Å), a carbon-hydrogen bond with LYS98 (3.34 Å), and a pi-donor hydrogen bond with THR101 (3.92 Å). These hydrogen bonds play a crucial role in stabilizing the complex by providing directional and specific interactions between the ligand and the protein.
In addition to hydrogen bonds, compound 2 also engages in hydrophobic interactions with three residues: THR101 (3.88 Å), LYS31 (5.18 Å), and PRO32 (4.38 Å). These interactions arise from the attraction between the non-polar regions of the ligand and the hydrophobic regions of the amino acid residues. They contribute to the overall binding affinity by providing non-polar contacts that minimize solvent exposure. Moreover, van der Waal's interactions, albeit weak, are also present between compound 1 and the protein. These interactions arise from the close proximity of the ligand and the protein, resulting in weak but additive attractions that further stabilize the complex.
Compound 2 forms diverse interactions with protein 4ZFI, encompassing hydrogen bonds, hydrophobic interactions, and van der Waal's interactions. These interactions collectively contribute to the high binding affinity, making compound 1 a potential therapeutic candidate for targeting 4ZFI.
Figure 12 illustrates the binding interactions between compound 2 and 4ZFI, revealing the molecular mechanisms governing their association. The 2D representation provides a planar view of the interactions, highlighting the amino acids involved in hydrogen bonding, hydrophobic interactions, and van der Waal's interactions. Conventional and carbon hydrogen bonds, depicted by green/sea green dashed lines, represent direct interactions between the ligand's functional groups and the protein's backbone or side chains. Pi-alkyl interactions, represented by pink dashed lines, signify non-polar interactions between the ligand's hydrophobic moieties and the protein's hydrophobic regions. Van der Waal's interactions, represented by light green lines, are weak but additive attractions between the ligand and the protein, arising from the close proximity of their electron clouds. These interactions collectively contribute to the overall binding affinity. The 3D ball and stick models provide a (3D) perspective of the binding interactions, allowing for a clearer understanding of the ligand's orientation within the protein's binding pocket. The ball model showcases the ligand and the protein residues in their respective shapes and sizes, while the stack model emphasizes the layering of the ligand and the protein residues within the binding pocket.
3.5 In-Silico Drug-likeness Predictions using SwissADME
Drug-likeness prediction involves assessing whether a given pharmacological agent possesses attributes consistent with effective oral drug administration (Agoni et al., 2023). Lipinski's rule outlines specific criteria for potential drug-like molecules, emphasizing attributes such as having fewer than five hydrogen-bond donors (HBDs), fewer than ten hydrogen-bond acceptors (HBAs), a molecular mass below 500 Da, a log P not exceeding five, and a total polar surface area (TPSA) not exceeding 140 Å (Baell et al., 2013).
Table 6
In-silico Pharmacokinetics Predictions of the Compounds (SwissADME)
| Parameters | Compound 1 | Compound 2 |
| Molecular Formula | C5H7N3O2 | C9H17F2NO3 |
| Molecular Weight (g/mol) | 141.13 | 219.19 |
| NHA | 3 | 6 |
| NHD | 2 | 4 |
| NRB | 1 | 3 |
| BS | 0.55 | 0.55 |
| SA | 1.49 | 2.41 |
| MLOGP | -1.04 | 0.90 |
| TPSA (A°2) | 87.25 | 72.72 |
| Log P (iLOGP) | 0.58 | 1.54 |
| Log S (ESOL) | -0.01 | -1.38 |
| Lipinski’s rule of five | 0 | 0 |
Keys: NBA: Number of H-bond acceptors; NHD: Number of H-bond donors; NRB: Number of Rotatable Bond; BA: Bioavailability Score; SA: Synthetic Accessibility, TPSA: Total Polar Surface Area, MLOGP: Molecular Logarithm of the Octanol-Water Partition Coefficient.
The findings from the current study indicate that all compounds adhere to Lipinski's rule of five (Table 6), suggesting their potential as candidates for anti-cancer investigations. The number of hydrogen bond acceptors (NHA), hydrogen bond donors (NHD), and rotatable bonds (NRB) are important factors in determining the drug-likeness of a compound. Lipinski's rule of five serves as a heuristic for anticipating a compound's drug-likeness. According to this rule, a compound is deemed drug-like if it possesses no more than five hydrogen bond donors (comprising nitrogen–hydrogen and oxygen–hydrogen bonds), no more than ten hydrogen bond acceptors (all derived from nitrogen or oxygen atoms), a molecular mass below 500 daltons, and a calculated octanol-water partition coefficient (Clog P) not surpassing 5 (Baell et al., 2013).
Compound 1 and Compound 2 exhibit promising drug-like properties based on their molecular characteristics and predicted values. Both compounds possess molecular weights within the desirable range for oral bioavailability, suggesting potential for effective absorption into the bloodstream. Their molecular formulas indicate relatively small and flexible structures, potentially facilitating distribution within the body. Moreover, both compounds exhibit the presence of rotatable bonds, allowing for conformational flexibility and potential interactions with biological targets.
Compound 1 demonstrates a moderate Bioavailability Score (BA) of 0.55, indicating a reasonable likelihood of absorption into the bloodstream. Its Synthetic Accessibility (SA) value of 1.49 suggests that the compound can be synthesized with relative ease, an important factor in drug development feasibility. The Molecular Logarithm of the Octanol-Water Partition Coefficient (MLOGP) value of -1.04 and Topological Polar Surface Area (TPSA) of 87.25 Ų suggest moderate lipophilicity and polar surface area, respectively, potentially influencing solubility and membrane permeability. While compound 2 also exhibits a moderate Bioavailability Score (BS) of 0.55, similar to Compound 1. Its Molecular Polar Surface Area (SA) of 2.41 suggests a limited polar region compared to Compound 1, potentially affecting solubility and absorption. The Molecular Logarithm of the Octanol-Water Partition Coefficient (MLOGP) value of 0.90 and Topological Polar Surface Area (TPSA) of 72.72 Ų suggest moderate lipophilicity and polarity, respectively. The Log P (iLOGP) value of 1.54 indicates a higher degree of hydrophobicity compared to Compound 1, potentially influencing membrane penetration. However, the Log S (ESOL) value of -1.38 suggests limited solubility in water compared to Compound 1.
Table 7
ADMET Profiles of the Compound 1 & 2
| Property | Parameter | Predicted value |
| Compound 1 | Compound 2 |
| Absorption (% Absorbed) | Human Intestinal Absorption | 71.335 | 82.383 |
| Distribution | BBB Permeability | -0.536 | -0.872 |
| CSN Permeability | -3.517 | -2.806 |
| Metabolism (Cytochrome P450, CYP) | CYP2D6 Substrate | No | No |
| CYP3A4 Substrate | No | No |
| CYP1A2 Inhibitor | No | No |
| CYP2C19 Inhibitor | No | No |
| CYP2C9 Inhibitor | No | No |
| CYP2D6 Inhibitor | No | No |
| CYP3A4 Inhibitor | No | No |
| Excretion | Total clearance | 0.728 | 0.698 |
| Toxicity | AMES Toxicity | Yes | Yes |
| Human Max. tolerated dose (log mg/kg/day) | 0.728 | 0.269 |
Table 7, the predicted values for absorption, distribution, metabolism, excretion (ADME), and toxicity properties for Compound 1 and Compound 2 provide insights into their pharmacokinetic and safety profiles. In terms of absorption, both compounds show high human intestinal absorption, with Compound 2 having a slightly higher percentage absorbed (82.383%) compared to Compound 1 (71.335%). Regarding distribution, the blood-brain barrier (BBB) permeability values for both compounds are negative, indicating limited penetration into the central nervous system (CNS). The cytochrome P450 (CYP) metabolism analysis reveals that neither compound is a substrate or inhibitor for major CYP enzymes (2D6, 3A4, 1A2, 2C19, 2C9), suggesting a lower likelihood of drug-drug interactions. In terms of excretion, both compounds exhibit comparable total clearance values, with Compound 1 at 0.728 and Compound 2 at 0.698. This suggests that both compounds have relatively efficient clearance rates from the body. However, in terms of toxicity, both compounds show AMES toxicity, indicating a potential to cause genetic mutations. The predicted human maximum tolerated dose, represented by the log mg/kg/day, is higher for Compound 1 (0.728) compared to Compound 2 (0.269), suggesting a potentially higher tolerance for Compound 1.
{kind=link}