4.1. Chemistry
All the chemicals, solvents and reagents used here without further purification, unless stated otherwise. The chemical reactions were monitored by Merk thin-layer chromatography (TLC) of silica gel 60 F254 aluminum plates. TLC plates were visualized under UV light at 254 nm using (UV visualizer name, company). For the concentration of organic solvents, Buchi rotary evaporator was used. Compounds were purified using normal phase column chromatography on silica gel (Merk Kieselgel 60, No. 9385, 230-400 mesh ASTM). 1H and 13C spectra were performed using Bruker DRX-500 spectrometer (operated at 300 MHz) and 500 MHz NMR instruments respectively, CDCl3 with tetramethylsilane used as internal standard, CD3OD and d6-DMSO were also used as d-solvents. Chemical shifts (δ) are expressed in parts per million (ppm) scale, reference to the residual solvent peaks (CDCl3: 1H 7.26 and 13C 77.16, CD3OD: 1H 3.31 and 13C 49.00, d6-DMSO: 1H 2.50 and 13C 39.52 and d6-Acetone: 1H 2.05 and 13C 29.84, 206.26 etc. Signal multiplicity are expressed as: br.s (broad singlet), s (singlet), d (doublet), t (triplet), q (quartet), p (pentet) and m (multiplet). To process NMR spectra, topspin software was used. Coupling constant (J) values are calculated in hertz (Hz). Melting point of all final compounds measured using Fargo MP-2D apparatus and are uncorrected. The high-resolution mass spectra were measured by JEOL (JMS-700) electron impact (EI) mass spectrometer. All the final compounds were checked (>95 %) and determined by HPLC (Agilent 1260 Infinity II, Agilent Technologies, Germany) using a Dikma (Diamonsil 5 µm C18x150x4.6 mm) column. HPLC analysis conditions used ACN (mobile phase A) and water containing NH4OAc 10 mM with HCOOH 0.1% as a solvent system (mobile phase B) with a flow rate of 0.5 mL/min.
4.1.1. Methyl 5-methyl-[1,1'-biphenyl]-2-carboxylate (19)
The synthetic procedure of compound 19 was conducted by following a procedure by Jeffrey et al. A mixture of methyl 2-bromobenzoate (6.5 g, 28.4 mmol), phenylboronic acid (5.36 g, 43.9 mmol), Pd(PPh3)4 (1.314 g, 3.68 mmol), K2CO3 (10.64 g, 71.0 mmol) in anhydrous DMF (65 mL) was heated at 110°C for 24 h under argon (Ar) atmosphere. The reaction mixture was cooled to room temperature, added with water then extracted with EA. The organic layer was combined, dried over anhydrous magnesium sulfate, and then concentrated under reduced pressure to obtain the residue. The resulting residue was purified by silica column chromatography (n-hexane: EA = 100: 1) to give compound 19 in 97% yield. Compound 19: 1H NMR (300 MHz, Chloroform-d) δH 7.77 (d, J = 7.8 Hz, 1H), 7.40 – 7.34 (m, 3H), 7.32 – 7.29 (m, 2H), 7.24 – 7.19 (m, 2H), 3.63 (s, 3H), 2.42 (s, 3H).
4.1.2. Methyl 5-(bromomethyl)-[1,1'-biphenyl]-2-carboxylate (20)
Adapting a procedure by Jeffrey et al., mixture of compound 19 (2.26 g, 10.0 mmol), N-bromosuccinimide (1.87 g, 10.5 mmol), 1,1′-azobis(cyclohexanecarbonitrile) (30 mg, 0.12 mmol) in carbon tetrachloride (40 mL) was refluxed at 85-90°C under argon for 23 h. The reaction mixture was cooled to room temperature, then extracted with EA. The organic layer was combined, dried over magnesium sulfate, and then concentrated under reduced pressure to obtain the residue. The resulting residue was purified by silica column chromatography (n-hexane: 100 à n-hexane: EA = 250:1 à 100: 1) to give compound 20 in 83% yield. Compound 20: 1H NMR (300 MHz, Chloroform-d) δH 7.81 (d, J = 7.8 Hz, 1H), 7.45 – 7.36 (m, 5H), 7.34 – 7.26 (m, 2H), 4.51 (s, 2H), 3.64 (s, 3H).
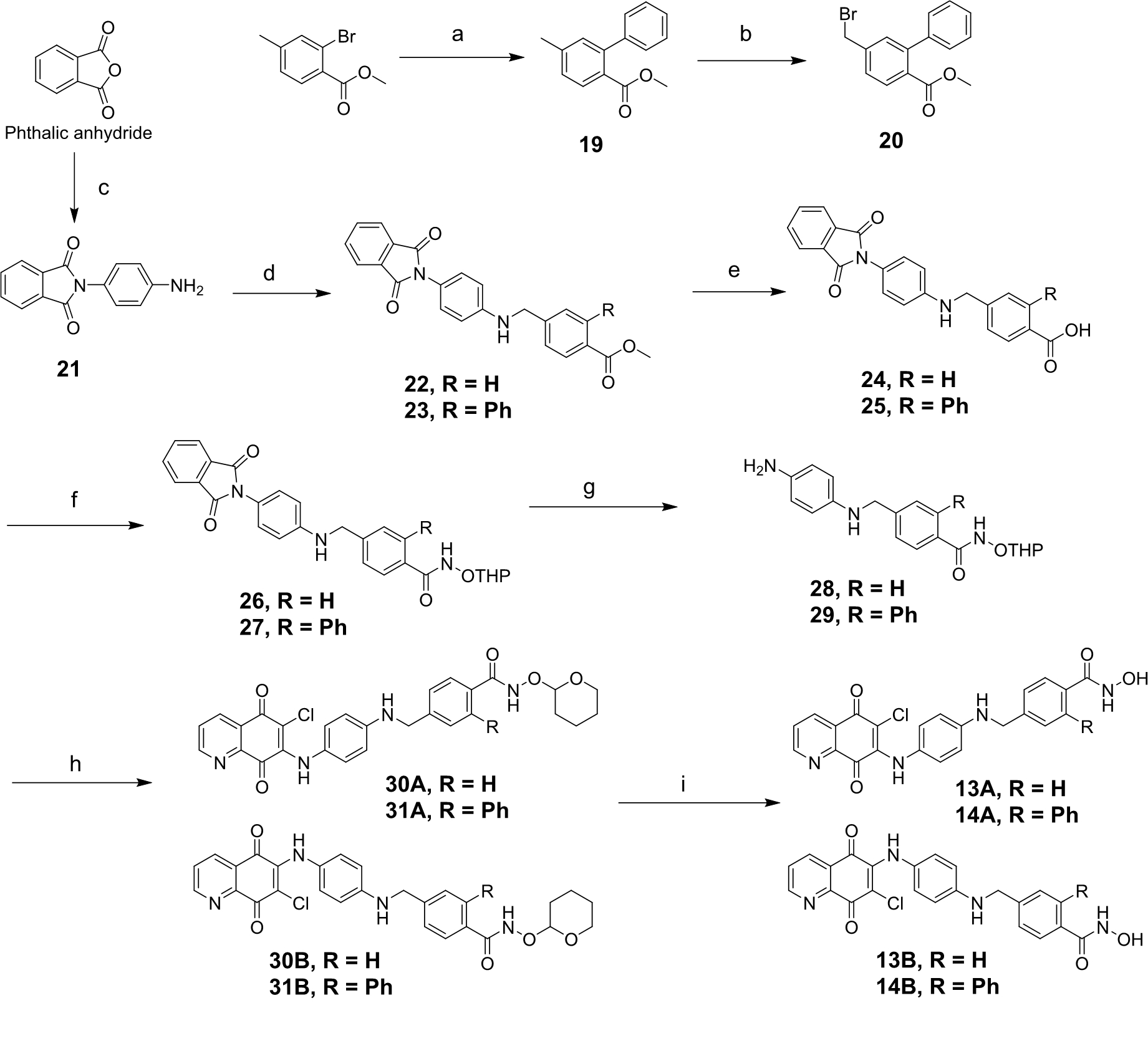
4.1.3. 2-(4-aminophenyl)isoindoline-1,3-dione (21)
A mixture of phthalic anhydride (14.812 g, 10 mmol) was dissolved in DMF (7 mL), and p-phenylenediamine (10.814 g, 10 mmol) was added in batches. The mixture was stirred under reflux at 150 oC and maintained for an overnight reaction. The reaction was monitored by TLC, after completion of the reaction, ice/water was poured into it, a green solid was precipitated, and collected after suction filtration to obtain compound 21 (65% yield). Compound 21: 1H NMR (300 MHz, DMSO-d6) δH 7.92 – 7.85 (m, 4H), 7.02 – 6.99 (m, 2H), 6.64 – 6.61 (m, 2H), 5.32 (s, 2H).
4.1.4. Methyl 4-(((4-(1,3-dioxoisoindolin-2-yl)phenyl)amino)methyl)benzoate (22)
A mixture of compound 21 (238 mg, 1 mmol), methyl 4-(bromomethyl)benzoate (229 mg, 1 mmol), and K2CO3 (138 mg, 1 mmol) in anhydrous DMF (3 mL) was stirred at room temperature for 24 h. After confirming the completion of the reaction using TLC, the mixture was quenched with water (5 mL). Then EA (10 mL) was added, and an orange solid was precipitated, which was filtered using suction to obtain compound 22 (67.3% yield). Compound 22: 1H NMR (300 MHz, DMSO-d6) δH 7.94 – 7.84 (m, 6H), 7.51 (d, J = 8.1 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 6.64 (t, J = 7.2 Hz, 1H), 6.62 (d, J = 7.2 Hz, 2H), 4.40 (d, J = 6.0 Hz, 2H), 3.82 (s, 3H).
4.1.5. Methyl 5-(((4-(1,3-dioxoisoindolin-2-yl) phenyl) amino) methyl)-[1,1'-biphenyl]-2- carboxylate (23)
A mixture of compound 21 (955 mg, 4 mmol), 20 (1220 mg, 4 mmol), and K2CO3 (552 mg, 4 mmol) in anhydrous DMF (10 mL) was stirred at room temperature for 24 h. TLC was monitored, and after confirming the reaction completion, the mixture was quenched with water (20 mL). Then EA (20 mL) was added, and a white solid was precipitated, which was filtered using suction to obtain compound 23 (56.4% yield). Compound 23: 1H NMR (300 MHz, DMSO-d6) δH 7.92 – 7.84 (m, 4H), 7.72 (d, J = 7.8 Hz, 1H), 7.48 – 7.35 (m, 5H), 7.28 – 7.7.26 (m, 2H), 7.08 – 7.05 (m, 2H), 6.68 – 6.65 (m, 3H), 4.41 (d, J = 6.3 Hz, 2H), 3.55 (s, 3H).
4.1.6. 4-(((4-(1,3-dioxoisoindolin-2-yl)phenyl)amino)methyl)benzoic acid (24) and 5-(((4-(1,3-dioxoisoindolin-2-yl)phenyl)amino)methyl)-[1,1'-biphenyl]-2-carboxylic acid (25)
To a solution of compound 22 or 23 (2 mmol) in MeOH (8 mL) was added lithium hydroxide 1.0 M (aq) (7 mL), and the reaction mixture was heated from room temperature to 55 oC and stirred for overnight. The solvent was reduced by half using a rotary evaporator to obtain a residue. The resulting residue was acidified using HCl 5% under an ice bath to adjust pH 4-5 and then EA was added, a brown solid precipitate was collected after suction filtration to obtain the desired product 24 or 25 respectively (85-90 % yield). The compound was directly used for the next steps without purification.
Compound 24: 1H NMR (300 MHz, DMSO-d6) δH 12.87 (br.s, 1H), 9.91 (s, 1H), 7.90 – 7.87 (m, 2H), 7.82 – 7.79 (m, 1H), 7.63 – 7.45 (m, 5H), 7.36 (d, J = 9.0 Hz, 2H), 6.55 (d, J = 8.7 Hz, 2H), 4.34 (s, 2H).
Compound 25: 1H NMR (300 MHz, DMSO-d6) δH 7.99 (d, J = 8.1 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.63 – 7.51 (m, 3H), 7.44 – 7.29 (m, 10H), 6.65 – 6.62 (m, 2H), 4.41 (s, 2H).
4.1.7. 4-(((4-(1,3-dioxoisoindolin-2-yl)phenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (26) and 5-(((4-(1,3-dioxoisoindolin-2-yl)phenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)-[1,1'-biphenyl]-2-carboxamide (27)
A mixture of 24 (500 mg, 1.34 mmol, 1 equv.) or 25 (580 mg, 1.29 mmol, 1 equv.), O-(tetrahydro-2H-pyran-3-yl)hydroxylamine (1.2 equv.), EDC. HCl (1.5 equv.), HOBt (1.2 equv), and N-methylmorpholine (1.5 equv.) in anhydrous DMF (5 mL) were stirred at room temperature for 3-4 h. The reaction mixture was quenched with water and extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA: MeOH = 2: 2: 0.1) to give the desired compound 26 or 27 respectively with 55-60% yield.
Compound 26: 1H NMR (300 MHz, DMSO-d6) δH 11.54 (s, 1H), 7.92 – 7.84 (m, 4H), 7.71 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.05 – 7.02 (m, 2H), 6.68 – 6.59 (m, 3H), 4.97 (s, 1H), 4.37 (d, J = 6.0 Hz, 2H), 4.05 – 3.99 (m, 1H), 3.52 – 3.48 (m, 1H), 1.70 (br.s, 3H), 1.53 (br.s, 3H).
Compound 27: 1H NMR (300 MHz, CDCl3) δH 8.09 (s, 1H), 7.92 – 7.90 (m, 2H), 7.77 – 7.74 (m, 2H), 7.65 (d, J = 7.8 Hz, 1H), 7.44 – 7.39 (m, 8H), 7.21 – 7.18 (m, 2H), 6.74 (d, J = 8.7 Hz, 2H), 4.75 (s, 1H), 4.44 (s, 2H), 3.63 – 3.56 (m, 1H), 3.42 – 3.36 (m, 1H), 1.73 (br.s, 3H), 1.49 (br.s, 3H)
4.1.8. 4-(((4-aminophenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (28) and 5-(((4-aminophenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)-[1,1'-biphenyl]-2-carboxamide (29)
A solution of 26 or 27 (1 mmol, 1 equv), and hydrazine hydrate (7 equv) in ethanol (10 mL) was stirred at 65-70 oC for 2-3 h under argon (Ar) gas. The resultant precipitate was filtered off and washed several times with ethanol, and the filtrate was collected and dried under vacuum to obtain the desired product 28 or 29 respectively with 65-70% yield.
Compound 28: 1H NMR (300 MHz, CDCl3) δH 9.02 (br.s, 1H), 7.69 (d, J = 8.1 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 6.60 – 6.55 (m, 2H), 6.50 – 6.45 (m, 2H), 5.06 (s, 1H), 4.30 (s, 2H), 4.03 – 3.96 (m, 1H), 3.67 – 3.60 (m, 1H), 3.21 (br.s, 2H), 1.86 (br.s, 3H), 1.60 (br.s, 3H).
Compound 29: 1H NMR (300 MHz, DMSO-d6) δH 11.78 (s, 1H), 7.79 – 7.70 (m, 8H), 6.77 (s, 4H), 5.87 (s, 1H), 5.27 (s, 1H), 4.63 (d, J = 5.7 Hz, 2H), 4.34 – 4.27 (m, 1H), 3.87 – 3.84 (m, 1H), 2.02 (br.s, 3H), 1.89 (br.s, 3H).
4.1.9. 4-(((4-((6-chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (30A) and 4-(((4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)benzamide (30B)
A mixture of 6,7-dichloroquinoline-5,8-dione (228 mg, 1 mmol), 28 (341 mg, 1 mmol), and DIPEA (175 µL, 1 mmol) in anhydrous DCM (9 mL) was stirred at room temperature for overnight. The reaction mixture was quenched with water (30 mL) and extracted using DCM (50 mL×2). The organic layer was separated, dried over anhydrous MgSO4 and evaporated in a vacuum. The residue was applied to silica column chromatography (n-hexane: EA: MeOH = 1: 1: 0.1) to give compounds 30A and 30B.
Compound 30A (purple solid, yield = 36 %): 1H NMR (300 MHz, CDCl3) δH 8.96 (dd, J = 4.8, 1.8 Hz, 1H), 8.78 (s, 1H), 8.50 (dd, J = 7.8, 1.8 Hz, 1H), 7.80 (s, 1H), 7.74 (d, J = 8.4, 2H), 7.68 (dd, J = 8.1, 4.8 Hz, 1H), 7.44 (d, J = 8.1, 2H), 6.95 (d, J = 8.4 Hz, 2H), 6.59 – 6.54 (m, 2H), 5.08 (t, J = 3.3, 1H), 4.42 (d, J = 4.2, 2H), 4.27 (s, 1H), 4.03 – 3.97 (m, 1H), 3.68 – 3.64 (m, 1H), 1.88 (br.s, 3H), 1.62 (br.s, 3H).
Compound 30B (purple solid, yield = 31 %): 1H NMR (300 MHz, Methanol-d4) δH 8.91 (dd, J = 4.8, 1.8 Hz, 1H), 8.46 (dd, J = 7.8, 1.8 Hz, 1H), 7.77 – 7.72 (m, 3H), 7.49 (d, J = 8.4, 2H 6.95 – 6.92 (m, 2H), 6.60 – 6.57 (m, 2H), 5.05 (br. s, 1H), 4.2 (s, 2H), 4.16 – 4.09 (m, 1H), 3.64 – 3.60 (m, 1H), 1.91 – 1.78 (m, 3H), 1.62 (br.s, 3H).
4.1.10. 5-(((4-((6-chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)-[1,1'-biphenyl]-2-carboxamide (31A) and 5-(((4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)amino)methyl)-N-((tetrahydro-2H-pyran-2-yl)oxy)-[1,1'-biphenyl]-2-carboxamide (31B)
Following the procedure for preparation of 30A and 30B, addition of 6,7-dichloroquinoline-5,8-dione (228 mg, 1 mmol), 29 (417 mg, 1 mmol), and DIPEA (175 µL, 1 mmol) in anhydrous DCM (9 mL) was stirred at room temperature for overnight, which gave compound 31A and 31B.
Compound 31A (purple solid, yield = 43%): 1H NMR (300 MHz, CDCl3) δH 8.95 (dd, J = 4.8, 1.8 Hz, 1H), 8.49 (dd, J = 7.8, 1.8 Hz, 1H), 7.99 (s, 1H), 7.81 (s, 1H), 7.70 – 7.66 (m, 2H), 7.43 – 7.38 (m, 7H), 6.95 (d, J = 8.7, 2H), 6.58 (d, J = 8.7 Hz, 2H), 4.75 (s, 1H), 4.43 (s, 2H), 4.31 (br. s, 1H), 3.58 – 3.54 (m, 1H), 3.40 – 3.36 (m, 1H), 1.73 (br.s, 3H), 1.49 (br.s, 3H).
Compound 31B (purple solid, yield = 41%): 1H NMR (300 MHz, CDCl3) δH 9.04 (dd, J = 4.5, 1.8 Hz, 1H), 8.40 (dd, J = 7.8, 1.8 Hz, 1H), 7.96 (s, 1H), 7.69 (d, J = 7.5 Hz, 1H), 7.63 – 7.59 (m, 2H), 7.44 – 7.39 (m, 7H), 6.95 (d, J = 8.7, 2H), 6.58 (d, J = 8.7 Hz, 2H), 4.75 (s, 1H), 4.44 (s, 2H), 4.30 (br. s, 1H), 3.61 – 3.54 (m, 1H), 3.40 – 3.36 (m, 1H), 1.74 (br.s, 3H), 1.50 (br.s, 3H).
4.1.11. 4-(((4-((6-chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)amino)methyl)-N-hydroxybenzamide (13A)
To a mixture of 30A (190 mg, 0.36 mmol) in dry THF (4 mL) and MeOH (2 mL) was added hydrochloric acid (0.5 mL) under an ice bath, stirred for 2 h, and then allowed to stir at room temperature for 30 min. The solvent was removed by using a rotary evaporator to obtain a crude product, and then dried under vacuum. The crude product was added DCM (30 mL) and a small amount of methanol (5 mL) to afford a precipitate. The precipitate was then washed with DCM and ether to give compound 13A.
Compound 13A: m.p. = 192-193 °C, purple solid, yield = 84%; 1H NMR (300 MHz, DMSO-d6) δH 11.21 (br. s, 1H), 9.32 (s, 1H), 8.92 (dd, J = 4.8, 1.8 Hz, 1H), 8.35 (dd, J = 7.8, 1.8 Hz, 1H), 7.81 (dd, J = 7.8, 4.8 Hz, 1H), 7.70 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 6.96 (d, J = 8.7, 2H), 6.75 (d, J = 8.7 Hz, 2H), 4.39 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δC 192.9, 178.4, 175.9, 163.8, 153.2, 146.6, 144.0, 133.9, 132.0, 129.5, 129.3, 128.6, 128.4, 127.6, 126.9, 125.2, 124.7, 122.9, 49.3. HRMS (ESI) calcd for C23H18O4N4Cl [M+H]+ 449.1011; found 449.1012. HPLC purity = 99.5% (tr = 13.821 min.).
4.1.12. 4-(((4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)amino)methyl)-N-hydroxybenzamide (13B)
Following the procedure for preparation of 13A, a mixture of 30B (160 mg, 0.30 mmol) in dry THF (4 mL) and MeOH (2 mL) followed by the addition of hydrochloric acid (0.5 mL) and stirred for 3 h under an ice-bath to get the desired compound 13B.
Compound 13B: m.p. = 180-181 °C, purple solid, yield = 90%; 1H NMR (300 MHz, DMSO-d6) δH 11.19 (br. s, 1H), 9.23 (s, 1H), 8.96 (dd, J = 4.8, 1.8 Hz, 1H), 8.35 (dd, J = 7.8, 1.8 Hz, 1H), 7.76 (dd, J = 7.8, 4.8 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 6.96 (d, J = 8.4, 2H), 6.75 (d, J = 7.2 Hz, 2H), 4.39 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δC 192.9, 179.9, 175.1, 163.8, 154.3, 147.7, 142.8, 134.6, 129.5, 128.7, 127.7, 127.5, 127.2, 126.9, 125.1, 124.6, 122.9, 54.9. HRMS (ESI) calcd for C23H18O4N4Cl [M+H]+ 449.1011; found 449.1012. HPLC purity = 99.3% (tr = 13.316 min.).
4.1.13. 5-(((4-((6-chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)amino)methyl)-N-hydroxy-[1,1'-biphenyl]-2-carboxamide (14A)
Following the procedure for preparation of 13A, a mixture of 31A (260 mg, 0.43 mmol) in dry THF (4 mL) and MeOH (2 mL) followed by the addition of hydrochloric acid (0.5 mL) and stirred for 2 h under an ice-bath to get the desired compound 14A.
Compound 14A: m.p. = 159-160 °C, purple solid, yield = 92%; 1H NMR (300 MHz, DMSO-d6) δH 10.75 (br. s, 1H), 9.20 (br. s, 1H), 8.92 – 8.32 (m, 2H), 8.33 (dd, J = 7.8, 1.8 Hz, 1H), 7.80 (dd, J = 7.8, 1.5 Hz, 1H), 7.39 – 7.31 (m, 8H), 6.88 (d, J = 8.7, 2H), 6.54 (d, J = 8.7 Hz, 2H), 6.46 (t, J = 6.3 Hz, 1H), 4.35 (d, J = 6.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δC 178.5, 175.5, 166.1, 153.0, 146.4, 146.3, 144.1, 142.0, 140.2, 139.6, 133.8, 132.8, 129.5, 128.8, 128.4, 128.3, 128.2, 127.4, 127.2, 126.0, 125.7, 111.5, 110.4, 46.1. HRMS (ESI) calcd for C29H22O4N4Cl [M+H]+ 525.1324; found 525.1327. HPLC purity = 96.2% (tr = 16.199 min.).
4.1.14. 5-(((4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)amino)methyl)-N-hydroxy-[1,1'-biphenyl]-2-carboxamide (14B)
Following the procedure for preparation of 13A, a mixture of 31B (250 mg, 0.41 mmol) in dry THF (4 mL) and MeOH (2 mL) followed by the addition of hydrochloric acid (0.5 mL) and stirred for 3 h under an ice-bath to get the desired compound 14B.
Compound 14B: m.p. = 220-221°C, purple solid, yield = 94%; 1H NMR (300 MHz, DMSO-d6) δH 10.75 (br. s, 1H), 9.15 (br. s, 1H), 8.97 – 8.93 (m, 2H), 8.34 (dd, J = 7.8, 1.8 Hz, 1H), 7.75 (dd, J = 7.8, 4.8 Hz, 1H), 7.40 – 7.32 (m, 8H), 6.89 (d, J = 8.7, 2H), 6.55 (d, J = 9.0 Hz, 2H), 6.44 (t, J = 6.3 Hz, 1H), 4.36 (d, J = 6.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δC 192.9, 179.8, 175.1, 165.8, 165.1, 154.3, 147.7, 139.8, 139.6, 134.6, 131.4, 131.1, 129.3, 128.5, 128.4, 128.3, 128.2, 127.8, 127.6, 124.8, 124.6, 123.0, 116.7, 114.3, 54.9. HRMS (ESI) calcd for C29H22O4N4Cl [M+H]+ 525.1324; found 525.1325. HPLC purity = 97.7% (tr = 15.689 min.).
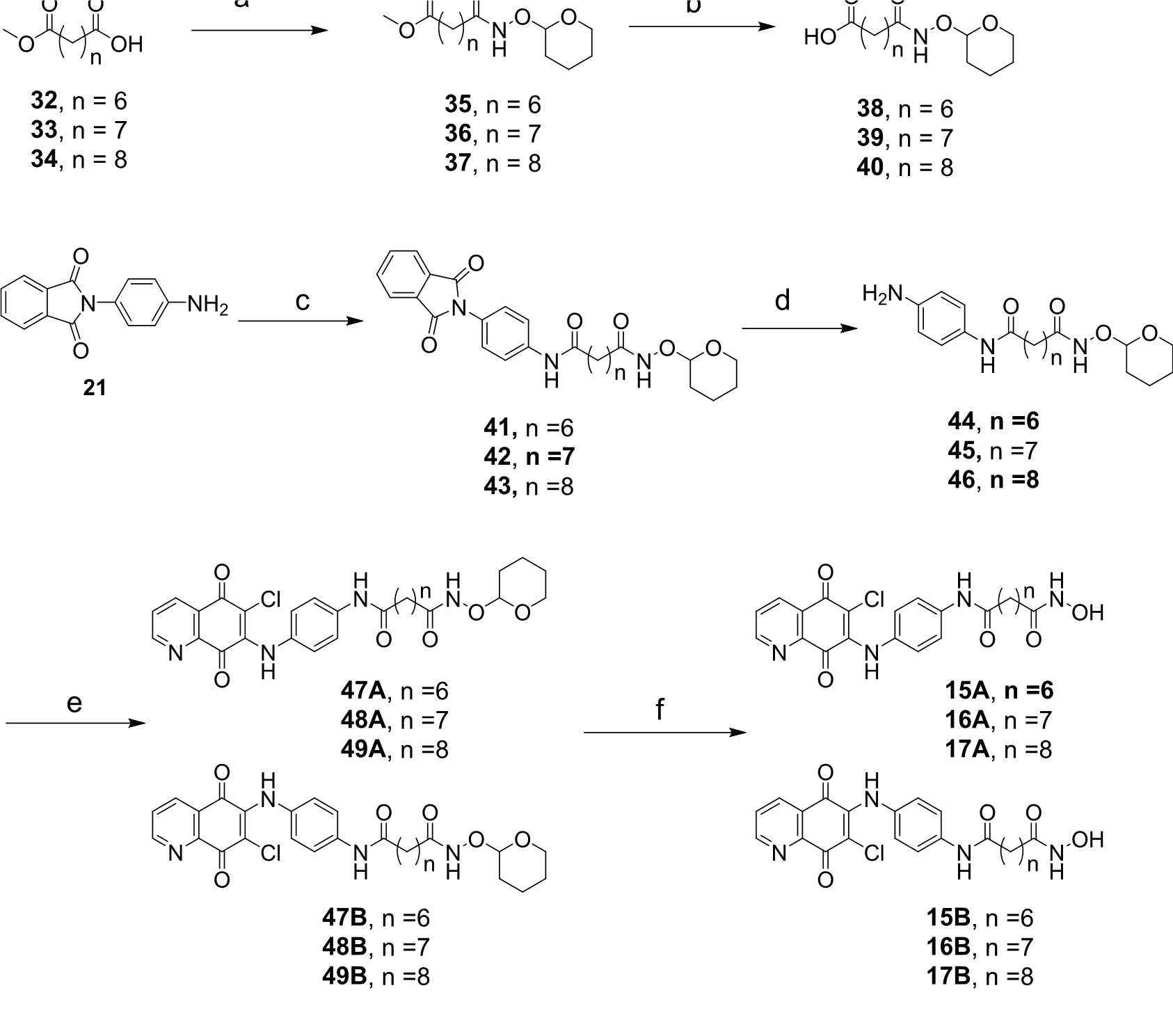
4.1.15. 8-Oxo-8-(((tetrahydro-2H-pyran-3-yl)oxy)amino)octanoic acid (38)
A mixture of monomethyl suberate 32 (941 mg, 5.0 mmol), O-(tetrahydro-2H-pyran-3-yl) hydroxylamine (879 mg, 7.5 mmol), EDC. HCl (1.43 g, 7.5 mmol), HOBt (811 mg, 6.0 mmol), and N-methylmorpholine (1.38 mL, 12.5 mmol) in anhydrous DMF (5 mL) was stirred at room temperature overnight. The reaction mixture was quenched with water and extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA = 2: 1) to give compound 35 with 90% yield. Compound 35: 1H NMR (300 MHz, Chloroform-d) δH 8.63 (s, 1H), 4.92 (br s, 1H), 3.99 – 3.79 (m, 1H), 3.64 (s, 3H), 3.63 – 3.56 (m, 1H), 2.28 (t, J = 7.5 Hz, 2H), 2.09 (br s, 2H), 1.86 – 1.73 (m, 3H), 1.67 – 1.54 (m, 7H), 1.38 – 1.27 (m, 4H).
To a solution of compound 35 (1.0 g, 3.48 mmol) in (15 mL) was added lithium hydroxide 1.0 M (aq) (10 mL), and the reaction mixture was stirred at heated at 50°C overnight. The solvent was removed by using a rotary evaporator to obtain a residue. The resulting residue was acidified using HCl 5% under an ice bath and then extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated by using a rotary evaporator to give compound 38 in 98% yield. The compound was directly used for further steps without purification. Compound 38: 1H NMR (300 MHz, DMSO-d6) δH 11.96 (s, 1H), 10.87 (s, 1H), 4.79 (br s, 1H), 3.99 – 3.83 (m, 1H), 3.57 – 3.43 (m, 1H), 2.18 (t, J = 7.4 Hz, 2H), 1.96 (t, J = 7.3 Hz, 2H), 1.72 – 1.57 (m, 3H), 1.56 – 1.41 (m, 7H), 1.30 – 1.19 (m, 4H).
4.1.16. 9-Oxo-9-(((tetrahydro-2H-pyran-3-yl)oxy)amino)nonanoic acid (39)
A mixture of monomethyl azelate 33 (1.7 g, 8.41 mmol), O-(tetrahydro-2H-pyran-3-yl)hydroxylamine (985 mg, 8.41 mmol), EDC. HCl (2.418 g, 12.6 mmol), HOBt (1.364 mg, 10.1 mmol), and N-methylmorpholine (2.31 mL, 21.0 mmol) in anhydrous DMF (7 mL) were stirred at room temperature overnight. The reaction mixture was quenched with water and extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA = 2: 1) to give compound 36 with 98% yield. Compound 36: 1H NMR (300 MHz, Chloroform-d) δH 8.72 (s, 1H), 4.91 (s, 1H), 4.04 – 3.86 (m, 1H), 3.63 (s, 3H), 3.62 – 3.52 (m, 1H), 2.28 (t, J = 7.4 Hz, 3H), 2.08 (br s, 2H), 1.82 – 1.74 (m, 3H), 1.64 – 1.53 (m, 7H), 1.33 – 1.26 (m, 6H).
To a solution of compound 36 (2.483 g, 8.24 mmol) in MeOH (10 mL) was added lithium hydroxide 1.0 M (aq) (20 mL), and the reaction mixture was stirred at heated at 50°C overnight. The solvent was removed by using a rotary evaporator to obtain a residue. The resulting residue was acidified using HCl 5% under an ice bath and then extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated by using a rotary evaporator to give compound 39 in 97% yield. The compound was directly used for further steps without purification. Compound 39: 1H NMR (300 MHz, Chloroform-d) δH 4.93 (br s, 1H), 4.05 – 3.84 (m, 1H), 3.76 – 3.56 (m, 1H), 2.34 (t, J = 7.4 Hz, 2H), 2.11 (br s, 2H), 1.87 – 1.74 (m, 3H), 1.68 – 1.57 (m, 7H), 1.36 – 1.30 (m, 6H).
4.1.17. 10-Oxo-10-(((tetrahydro-2H-pyran-3-yl)oxy)amino)decanoic acid (40)
A mixture of monomethyl sebacate 34 (850 mg, 3.93 mmol), O-(tetrahydro-2H-pyran-3-yl)hydroxylamine (460 mg, 3.93 mmol), EDC. HCl (1.13 g, 5.89 mmol), HOBt (638 mg, 4.72 mmol), and N-methylmorpholine (1.08 mL, 9.82 mmol) in anhydrous DMF (5 mL) was stirred at room temperature overnight. The reaction mixture was quenched with water and extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA = 2: 1) to give compound 37 with 83% yield. Compound 37: 1H NMR (300 MHz, Chloroform-d) δH 4.91 (br s, 1H), 3.98 – 3.86 (m, 1H), 3.64 (s, 3H), 3.61 – 3.54 (m, 1H), 2.27 (t, J = 7.5 Hz, 2H), 2.08 (br s, 2H), 1.85 – 1.73 (m, 3H), 1.69 – 1.53 (m, 7H), 1.30 – 1.23 (m, 8H).
To a solution of compound 37 (1.07 g, 3.93 mmol) in dioxane (15 mL) was added lithium hydroxide 1.0 M (aq) (7 mL), and the reaction mixture was stirred at room temperature for overnight. The solvent was removed by using a rotary evaporator to obtain a residue. The resulting residue was acidified using HCl 5% under an ice bath and then extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated by using a rotary evaporator to give compound 40 in 98% yield. The compound was directly used for further steps without purification. Compound 40: 1H NMR (300 MHz, Chloroform-d) δH 4.92 (br s, 1H), 4.02 – 3.89 (m, 1H), 3.68 – 3.56 (m, 1H), 2.32 (t, J = 7.4 Hz, 2H), 2.14 (br s, 2H), 1.85 – 1.74 (m, 3H), 1.69 – 1.58 (m, 7H), 1.33 – 1.27 (m, 8H).
4.1.18. N1-(4-(1,3-dioxoisoindolin-2-yl)phenyl)-N8-((tetrahydro-2H-pyran-3-yl)oxy)octanediamide (41)
A mixture of 8-oxo-8-(((tetrahydro-2H-pyran-3-yl)oxy)amino)octanoic acid 38 (550 mg, 2.01 mmol), 2-(4-aminophenyl)isoindoline-1,3-dione 21 (400 mg, 1.68 mmol), EDC. HCl (480 mg, 2.50 mmol), HOBt (271 mg, 2.0 mmol), and N-methylmorpholine (460 µL, 4.17 mmol) in anhydrous DMF (5 mL) was stirred at room temperature overnight. The mixture was quenched with water, then added EA. The white-formed precipitate was filtered by vacuum filtration to obtain the crude product. The crude product was washed with n-hexane and EA to give compound 41 with 83% yield. Compound 41: 1H NMR (300 MHz, DMSO-d6) δH 10.89 (s, 1H), 10.05 (s, 1H), 8.07 – 7.83 (m, 4H), 7.83 – 7.68 (m, 2H), 7.47 – 7.31 (m, 2H), 4.81 (br s, 1H), 4.03 – 3.84 (m, 1H), 3.59 – 3.46 (m, 1H), 2.33 (t, J = 7.4 Hz, 2H), 2.00 (d, J = 7.3 Hz, 1H), 1.76 – 1.45 (m, 10H), 1.41 – 1.21 (m, 4H).
4.1.19. N1-(4-(1,3-Dioxoisoindolin-2-yl)phenyl)-N9-((tetrahydro-2H-pyran-3-yl)oxy)nonanediamide (42)
The preparation of compound 42 is similar to that of compound 41 by using 9-oxo-9-(((tetrahydro-2H-pyran-3-yl)oxy)amino)nonanoic acid (39) (658 mg, 2.29 mmol), 2-(4-aminophenyl)isoindoline-1,3-dione (21) (694 mg, 2.91 mmol), EDC. HCl (659 mg, 3.44 mmol), HOBt (372 mg, 2.75 mmol), and N-methylmorpholine (629 µL, 5.72 mmol) in anhydrous DMF (5 mL). Compound 42: 1H NMR (300 MHz, DMSO-d6) δH 10.88 (s, 1H), 10.05 (s, 1H), 7.98 – 7.89 (m, 4H), 7.75 – 7.67 (m, 2H), 7.40 – 7.30 (m, 2H), 4.79 (br s, 1H), 3.97 – 3.86 (m, 1H), 3.57 – 3.43 (m, 1H), 2.33 (t, J = 7.3 Hz, 2H), 1.98 (t, J = 7.5 Hz, 2H), 1.70 – 1.41 (m, 10H), 1.38 – 1.25 (m, 6H).
4.1.20. N1-(4-(1-Methylene-3-oxoisoindolin-2-yl)phenyl)-N10-((tetrahydro-2H-pyran-3-yl)oxy)decanediamide (43)
The preparation of compound 43 is similar to that of compound 41 by using 10-oxo-10-(((tetrahydro-2H-pyran-3-yl)oxy)amino)decanoic acid 40 (420 mg, 1.39 mmol), 2-(4-aminophenyl)isoindoline-1,3-dione 21 (365 mg, 1.53 mmol), EDC. HCl (401 mg, 2.09 mmol), HOBt (226 mg, 1.67 mmol), and N-methylmorpholine (383 µL, 3.48 mmol) in anhydrous DMF (5 mL). Compound 43: 1H NMR (300 MHz, DMSO-d6) δH 10.88 (s, 1H), 10.04 (s, 1H), 8.00 – 7.87 (m, 4H), 7.76 – 7.65 (m, 2H), 7.45 – 7.31 (m, 2H), 4.79 (br s, 1H), 4.05 – 3.79 (m, 1H), 3.61 – 3.44 (m, 1H), 2.33 (t, J = 7.4 Hz, 2H), 1.97 (t, J = 7.3 Hz, 2H), 1.73 – 1.39 (m, 10H), 1.35 – 1.23 (m, 8H).
4.1.21. N1-(4-Aminophenyl)-N8-((tetrahydro-2H-pyran-3-yl)oxy)octanediamide (44)
Compound 41 (508 mg, 1.0 mmol) and ethanol (10 mL) were placed in a round-bottom flask. Then, hydrazine 64% (253 µL, 5.0 mmol) was added to the reaction mixture. The flask was degassed and flushed with an argon gas. The reaction mixture was then heated at 50-60 °C for 4-5 h. The white precipitate was observed during this reaction. After confirming the completion of the reaction by TLC, the reaction was cooled to room temperature, then the white precipitate was filtered by vacuum filtration. The filtrate was collected and concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (DCM: MeOH = 4: 0.1) to give compound 44 in 70% yield. Compound 44: 1H NMR (300 MHz, DMSO-d6) δH 10.88 (s, 1H), 9.39 (s, 1H), 7.25 – 7.14 (m, 2H), 6.53 – 6.42 (m, 2H), 4.79 (s, 3H), 3.94 – 3.88 (m, 1H), 3.54 – 3.44 (m, 1H), 2.19 (t, J = 7.4 Hz, 2H), 1.97 (t, J = 7.3 Hz, 2H), 1.71 – 1.46 (m, 10H), 1.30 – 1.21 (m, 4H).
4.1.22. N1-(4-Aminophenyl)-N9-((tetrahydro-2H-pyran-3-yl)oxy)nonanediamide (45)
The preparation of compound 45 is similar to that of the compound 44 by using N1-(4-(1,3-dioxoisoindolin-2-yl)phenyl)-N9-((tetrahydro-2H-pyran-3-yl)oxy)nonanediamide 42 (451 mg, 0.89 mmol) and hydrazine 64% (269 μL, 5.33 mmol) in ethanol (10 mL). Purification was accomplished by silica column chromatography (DCM: MeOH = 3: 0.1) to give compound 45 in 91% yield. Compound 45: 1H NMR (300 MHz, DMSO-d6) δH 10.88 (s, 1H), 9.39 (s, 1H), 7.29 – 7.15 (m, 2H), 6.57 – 6.39 (m, 2H), 4.79 (s, 3H), 3.91 (s, 1H), 3.58 – 3.44 (m, 1H), 2.19 (t, J = 7.4 Hz, 2H), 1.97 (t, J = 7.2 Hz, 2H), 1.69 – 1.45 (m, 10H), 1.32 – 1.19 (m, 6H).
4.1.23. N1-(4-Aminophenyl)-N10-((tetrahydro-2H-pyran-3-yl)oxy)decanediamide (46)
The preparation of compound 46 is similar to that of the compound 44 by using N1-(4-(1-methylene-3-oxoisoindolin-2-yl)phenyl)-N10-((tetrahydro-2H-pyran-3-yl)oxy)decanediamide 43 (470 mg, 0.9 mmol), hydrazine 64% (270 μL, 5.4 mmol) in ethanol (10 mL). Purification was accomplished by silica column chromatography (DCM: MeOH = 5: 0.1) to give compound 46 in 97% yield. Compound 46: 1H NMR (300 MHz, DMSO-d6) δH 10.88 (s, 1H), 9.39 (s, 1H), 7.34 – 7.10 (m, 2H), 6.57 – 6.39 (m, 2H), 4.79 (s, 3H), 3.99 – 3.80 (m, 1H), 3.55 – 3.40 (m, 1H), 2.19 (t, J = 7.4 Hz, 2H), 1.96 (t, J = 7.2 Hz, 2H), 1.79 – 1.44 (m, 10H), 1.32 – 1.20 (m, 8H).
4.1.24. N1-(4-((6-Chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N8-((tetrahydro-2H-pyran-2-yl)oxy)octanediamide (47A) and N1-(4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N8-((tetrahydro-2H-pyran-2-yl)oxy)octanediamide (47B)
A mixture of 6,7-dichloroquinoline-5,8-dione (190 mg, 0.83 mmol), N1-(4-aminophenyl)-N8-((tetrahydro-2H-pyran-3-yl)oxy)octanediamide 44 (252 mg, 0.69 mmol), and DIPEA (133 µL, 0.76 mmol) in anhydrous DCM was stirred at room temperature overnight. The reaction mixture was concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatoraphy (n-hexane: EA: MeOH = 1: 2: 0.2) to give compounds 47A and 47B.
Compound 47A (purple solid, yield = 20%): 1H NMR (300 MHz, Methanol-d4) δH 8.90 (dd, J = 4.8, 1.7 Hz, 1H), 8.51 (dd, J = 7.9, 1.7 Hz, 1H), 7.83 (dd, J = 7.9, 4.8 Hz, 1H), 7.61 – 7.51 (m, 2H), 7.19 – 7.07 (m, 2H), 4.89 (br s, 1H), 4.06 – 3.93 (m, 1H), 3.65 – 3.55 (m, 1H), 2.38 (t, J = 7.4 Hz, 2H), 2.13 (t, J = 7.3 Hz, 2H), 1.81 – 1.56 (m, 10H), 1.46 – 1.37 (m, 4H).
Compound 47B (purple solid, yield = 20%): 1H NMR (300 MHz, DMSO-d6) δH 10.89 (s, 1H), 9.90 (s, 1H), 8.98 (dd, J = 4.8, 1.5 Hz, 1H), 8.37 (dd, J = 8.1, 1.3 Hz, 1H), 7.78 (dd, J = 7.9, 4.7 Hz, 1H), 7.61 – 7.49 (m, 2H), 7.24 – 6.93 (m, 2H), 4.80 (br s, 1H), 4.08 – 3.79 (m, 1H), 3.53 – 3.49 (m, 1H), 2.29 (t, J = 7.4 Hz, 2H), 1.98 (t, J = 7.3 Hz, 2H), 1.66 – 1.48 (m, 10H), 1.32 – 1.25 (m, 4H).
4.1.25. N1-(4-((6-Chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N9-((tetrahydro-2H-pyran-2-yl)oxy)nonanediamide (48A) and N1-(4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N9-((tetrahydro-2H-pyran-3-yl)oxy)nonanediamide (48B)
A mixture of 6,7-dichloroquinoline-5,8-dione (151 mg, 0.66 mmol), N1-(4-aminophenyl)-N9-((tetrahydro-2H-pyran-3-yl)oxy)nonanediamide 45 (167 mg, 0.45 mmol), and DIPEA (86 µL, 0.49 mmol) in anhydrous DCM was stirred at room temperature overnight. The reaction mixture was concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA: MeOH = 1: 2: 0.2) to give compounds 48A and 48B.
Compound 48A (purple solid, yield = 41%): 1H NMR (300 MHz, Methanol-d4) δH 8.90 (dd, J = 4.8, 1.7 Hz, 1H), 8.51 (dd, J = 7.9, 1.7 Hz, 1H), 7.83 (dd, J = 7.9, 4.8 Hz, 1H), 7.62 – 7.51 (m, 2H), 7.18 – 7.08 (m, 2H), 4.07 – 3.93 (m, 1H), 3.65 – 3.53 (m, 1H), 2.38 (t, J = 7.5 Hz, 2H), 2.12 (t, J = 7.3 Hz, 2H), 1.81 – 1.54 (m, 10H), 1.45 – 1.35 (m, 6H).
Compound 48B (purple solid, yield 40%): 1H NMR (300 MHz, Methanol-d4) 1H NMR (300 MHz, Methanol-d4) δH 8.93 (dd, J = 4.8, 1.7 Hz, 1H), 8.50 (dd, J = 7.9, 1.7 Hz, 1H), 7.78 (dd, J = 7.9, 4.8 Hz, 1H), 7.61 – 7.51 (m, 2H), 7.18 – 7.08 (m, 2H), 4.06 – 3.93 (m, 1H), 3.60 – 3.54 (m, 1H), 2.38 (t, J = 7.5 Hz, 2H), 2.12 (t, J = 7.4 Hz, 2H), 1.83 – 1.54 (m, 10H), 1.45 – 1.34 (m, 6H).
4.1.26. N1-(4-((6-Chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N10-((tetrahydro-2H-pyran-2-yl)oxy)decanediamide (49A) and N1-(4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N10-((tetrahydro-2H-pyran-2-yl)oxy)decanediamide (49B)
A mixture of 6,7-dichloroquinoline-5,8-dione (228 mg, 1.0 mmol), N1-(4-aminophenyl)-N10-((tetrahydro-2H-pyran-3-yl)oxy)decanediamide 46 (383 mg, 1.01 mmol), and DIPEA (192 µL, 1.11 mmol) in anhydrous THF was stirred at room temperature overnight. The reaction mixture was concentrated by using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA: MeOH = 1: 2: 0.2) to give compounds 49A and 49B.
Compound 49A (purple solid, yield = 42%): 1H NMR (300 MHz, Methanol-d4) δH 8.90 (dd, J = 4.8, 1.7 Hz, 1H), 8.51 (dd, J = 7.9, 1.7 Hz, 1H), 7.83 (dd, J = 7.9, 4.8 Hz, 1H), 7.63 – 7.50 (m, 2H), 7.18 – 7.08 (m, 2H), 4.88 (br s, 1H), 4.05 – 3.94 (m, 1H), 3.65 – 3.53 (m, 1H), 2.38 (t, J = 7.4 Hz, 2H), 2.11 (t, J = 7.4 Hz, 2H), 1.83 – 1.56 (m, 10H), 1.37 (s, 8H).
Compound 49B (purple solid, yield =40%): 1H NMR (300 MHz, DMSO-d6) δH 10.88 (s, 1H), 9.88 (s, 1H), 9.30 (br s, 1H), 8.98 (dd, J = 4.7, 1.6 Hz, 1H), 8.37 (dd, J = 7.8, 1.1 Hz, 1H), 7.78 (dd, J = 7.8, 4.7 Hz, 1H), 7.57 – 7.48 (m, 2H), 7.11 – 7.02 (m, 2H), 4.79 (br s, 1H), 3.94 – 3.88 (m, 1H), 3.53 – 3.43 (m, 1H), 2.29 (t, J = 7.3 Hz, 2H), 1.97 (t, J = 7.2 Hz, 2H), 1.73 – 1.42 (m, 10H), 1.34 – 1.22 (m, 8H).
4.1.27. N1-(4-((6-Chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N8-hydroxyoctanediamide (15A)
compound 47A (110 mg, 0.198 mmol) was dissolved in dry THF (4 mL) and MeOH (1 mL). While stirring under an ice bath, concentrated hydrochloric acid (0.6 mL) was added to it dropwise. The reaction mixture was stirred for 2-3 h. The solvent was removed by using a rotary evaporator to obtain the crude product, then it was purified by silica column chromatography (DCM: MeOH =3: 0.3 à 3: 0.4 à 3: 0.5) to give compound 15A.
Compound 15A: m.p. = 69-70 °C, purple solid, yield = 81%; 1H NMR (300 MHz, DMSO-d6) δH 10.36 (s, 1H), 9.98 (s, 1H), 8.95 (dd, J = 4.7, 1.7 Hz, 1H), 8.65 (s, 1H), 8.37 (dd, J = 7.9, 1.7 Hz, 1H), 7.83 (dd, J = 7.9, 4.7 Hz, 1H), 7.57 – 7.52 (m, 2H), 7.10 – 7.05 (m, 2H), 2.30 (t, J = 7.5 Hz, 2H), 1.94 (t, J = 7.3 Hz, 2H), 1.60 – 1.47 (m, 4H), 1.35 – 1.28 (m, 4H); 13C NMR (151 MHz, DMSO-d6) δC 178.4, 176.0, 171.2, 169.1, 153.2, 146.6, 144.0, 136.4, 133.9, 133.6, 129.7, 129.3, 128.4, 124.7, 118.5, 112.4, 36.3, 32.2, 28.4 (2C), 25.0 (2C). HRMS (ESI) calcd for C23H24O5N4Cl [M+H]+ 471.1435; found 471.1437. HPLC purity = 95.5% (tr = 12.607 min.).
4.1.28. N1-(4-((7-Chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N8-hydroxyoctanediamide (15B)
To a mixture N1-(4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N8-((tetrahydro-2H-pyran-2-yl)oxy)octanediamide 47B (320 mg, 0.58 mmol) in dry THF (3 mL) and MeOH (2 mL) was added hydrochloric acid (0.6 mL). The mixture was stirred under an ice bath for 2 h, and then allowed to stir at room temperature for 30 min. The solvent was removed by using a rotary evaporator to obtain a crude product, and then dried under vacuum. The crude product was added DCM and small amount of methanol to afford a precipitate. The precipitate was then washed with DCM and ether to give compound 15B.
Compound 15B: m.p. = 225-226 °C, purple solid, yield = 54%; 1H NMR (300 MHz, DMSO-d6) δH 10.32 (s, 1H), 9.89 (s, 1H), 9.32 (s, 1H), 8.98 (dd, J = 4.7, 1.7 Hz, 1H), 8.64 (s, 1H), 8.37 (dd, J = 7.9, 1.7 Hz, 1H), 7.78 (dd, J = 7.9, 4.7 Hz, 1H), 7.56 – 7.51 (m, 2H), 7.10 – 7.05 (m, 2H), 2.29 (t, J = 7.4 Hz, 2H), 1.94 (t, J = 7.3 Hz, 2H), 1.60 – 1.47 (m, 4H), 1.31 – 1.28 (m, 4H); 13C NMR (151 MHz, DMSO-d6) δC 179.9, 175.0, 171.2, 169.1, 154.4, 147.8, 142.9, 136.5, 134.4, 133.5, 127.5, 127.2, 124.6, 118.5, 114.4, 36.3, 32.2, 28.4 (2C), 25.0 (2C). HRMS (ESI) calcd for C23H24O5N4Cl [M+H]+ 471.1435; found 471.1431. HPLC purity = 95.3% (tr = 12.123 min.).
4.1.29. N1-(4-((6-Chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N9-hydroxynonanediamide (16A)
The synthesis method of 16A is similar to that of compound 15A by using N1-(4-((6-chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N9-((tetrahydro-2H-pyran-2-yl)oxy)nonanediamide 48A (105 mg, 0.185 mmol). The crude product was purified by silica column chromatography (DCM: MeOH =3: 0.3 à 3: 0.4 à 3: 0.5) to give compound 16A.
Compound 16A: m.p. = 184-185 °C, purple solid, yield = 92%; 1H NMR (300 MHz, DMSO-d6) δH 10.36 (br s, 1H), 9.99 (s, 1H), 9.24 (br s, 1H), 8.94 (dd, J = 4.7, 1.7 Hz, 1H), 8.65 (br s, 1H), 8.37 (dd, J = 7.8, 1.7 Hz, 1H), 7.83 (dd, J = 7.9, 4.7 Hz, 1H), 7.57 – 7.52 (m, 2H), 7.09 – 7.04 (m, 2H), 2.30 (t, J = 7.4 Hz, 2H), 1.94 (t, J = 7.3 Hz, 2H), 1.60 – 1.46 (m, 4H), 1.32 – 1.25 (m, 6H); 13C NMR (151 MHz, DMSO-d6) δC 178.4, 176.0, 171.2, 169.1, 153.2, 146.6, 144.1, 136.4, 133.9, 133.6, 129.3, 128.3, 124.7, 118.5, 112.4, 36.3, 32.2, 28.6, 28.5, 28.4, 25.1(2C). HRMS (ESI) calcd for C24H26O5N4Cl [M+H]+ 485.1592; found 485.1585. HPLC purity = 97.0% (tr = 13.443 min.).
4.1.30. N1-(4-((7-Chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N9-hydroxynonanediamide (16B)
The synthesis method of compound 16B is similar to that of compound 15A by using N1-(4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N9-((tetrahydro-2H-pyran-3-yl)oxy)nonanediamide 48B (171 mg, 0.30 mmol). The solvent was removed by using a rotary evaporator to obtain a crude product, and then dried under vacuum. The crude product was added DCM and small amount of methanol to afford a precipitate. The precipitate was then washed with DCM and ether to give compound 16B.
Compound 16B: m.p. = 222-223 °C, purple solid, yield = 42%; 1H NMR (300 MHz, DMSO-d6) δH 10.31 (s, 1H), 9.88 (s, 1H), 9.32 (s, 1H), 8.98 (dd, J = 4.7, 1.7 Hz, 1H), 8.62 (br s, 1H), 8.37 (dd, J = 7.8, 1.7 Hz, 1H), 7.78 (dd, J = 7.9, 4.7 Hz, 1H), 7.56 – 7.51 (m, 2H), 7.10 – 7.05 (m, 2H), 2.29 (t, J = 7.4 Hz, 2H), 1.94 (t, J = 7.3 Hz, 2H), 1.60 – 1.46 (m, 4H), 1.32 – 1.28 (m, 6H); 13C NMR (151 MHz, DMSO-d6) δC 180.0, 175.2, 171.1, 169.1, 154.4, 147.8, 142.8, 136.3, 134.4, 133.6, 127.5, 127.1, 124.7, 118.5, 114.5, 36.4, 32.2, 28.6, 28.5, 28.4, 25.1(2C). HRMS (ESI) calcd for C24H26O5N4Cl [M+H]+ 485.1592; found 485.1585. HPLC purity = 95.1% (tr = 15.340 min.).
4.1.31. N1-(4-((6-Chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N10-hydroxydecanediamide (17A)
The synthesis method of compound 17A is similar to that of compound 15A by using N1-(4-((6-chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)-N10-((tetrahydro-2H-pyran-2-yl)oxy)decanediamide 49A (250 mg, 0.43 mmol). The crude product was purified by silica column chromatography (DCM: MeOH = 3: 0.1 à 3: 0.2) to give compound 17A.
Compound 17A: m.p. = 190-191 °C, purple solid, yield = 95%; 1H NMR (300 MHz, DMSO-d6) δH 10.31 (s, 1H), 9.88 (s, 1H), 9.40 (br s, 1H), 8.94 (dd, J = 4.6, 1.6 Hz, 1H), 8.64 (s, 1H), 8.37 (dd, J = 7.9, 1.7 Hz, 1H), 7.83 (dd, J = 7.8, 4.7 Hz, 1H), 7.55 – 7.51 (m, 2H), 7.10 – 7.05 (m, 2H), 2.29 (t, J = 7.3 Hz, 2H), 1.93 (t, J = 7.3 Hz, 2H), 1.61 – 1.46 (m, 4H), 1.32 – 1.26 (m, 8H); 13C NMR (151 MHz, DMSO-d6) δC 178.4, 176.0, 171.1, 169.1, 153.2, 146.6, 144.1, 136.3, 133.9, 133.6, 129.3, 128.3, 124.7, 118.5, 112.4, 36.4, 32.2, 28.7(2C), 28.6(2C), 25.1(2C). HRMS (ESI) calcd for C25H28O5N4Cl [M+H]+ 499.1748; found 499.1748. HPLC purity = 97.5% (tr = 14.274 min.).
4.1.32. N1-(4-((7-Chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N10-hydroxydecanediamide (17B)
The synthesis method of compound 17B is similar to that of compound 15A by using N1-(4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)-N10-((tetrahydro-2H-pyran-2-yl)oxy)decanediamide 49B (230 mg, 0.39 mmol). The solvent was removed by using a rotary evaporator to obtain a crude product, and then dried under vacuum. The crude product was added DCM and a small amount of methanol to afford a precipitate. The precipitate was then washed with dichloromethane and ether to give compound 17B.
Compound 17B: m.p. = 215-216 °C, purple solid, yield = 47%; 1H NMR (300 MHz, DMSO-d6) δH 10.31 (s, 1H), 9.88 (s, 1H), 9.24 (br s, 1H), 8.98 (dd, J = 4.7, 1.7 Hz, 1H), 8.64 (s, 1H), 8.37 (dd, J = 7.8, 1.7 Hz, 1H), 7.78 (dd, J = 7.9, 4.7 Hz, 1H), 7.56 – 7.51 (m, 2H), 7.10 – 7.05 (m, 2H), 2.29 (t, J = 7.3 Hz, 2H), 1.93 (t, J = 7.3 Hz, 2H), 1.61 – 1.46 (m, 4H), 1.27 – 1.23 (m, 8H). 13C NMR (151 MHz, DMSO-d6) δC 180.0, 175.2, 171.2, 169.1, 154.4, 147.8, 142.9, 136.3, 134.4, 133.7, 129.7, 127.5, 124.7, 118.5, 114.5, 36.4, 32.3, 28.7(2C), 28.6(2C), 25.1(2C). HRMS (ESI) calcd for C25H28O5N4Cl [M+H]+ 499.1748; found 499.1741. HPLC purity = 95.2% (tr = 13.780 min.).
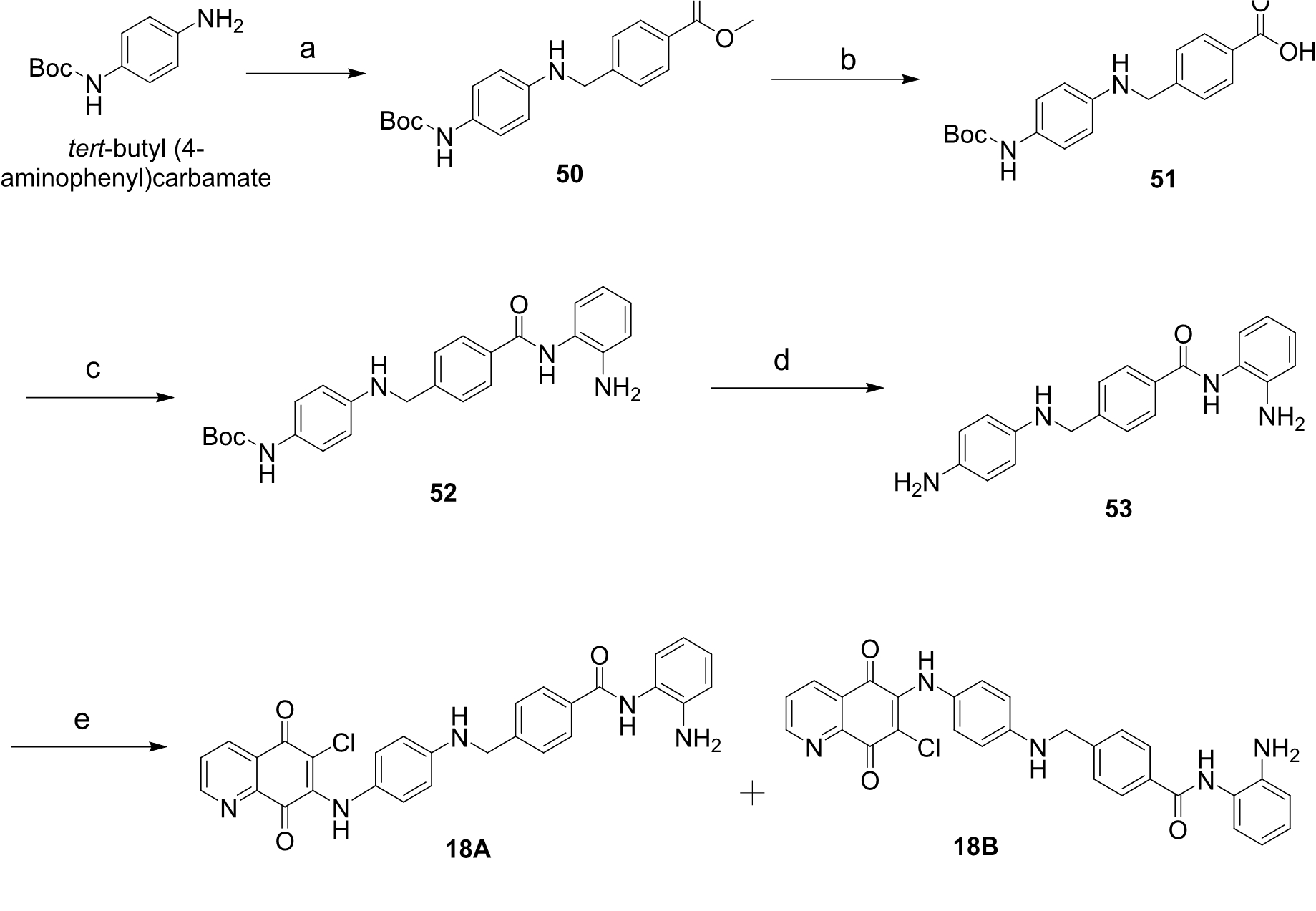
4.1.33. 4-(((4-((tert-butoxycarbonyl)amino)phenyl)amino)methyl)benzoic acid (51)
A mixture of methyl 4-(bromomethyl)benzoate (750 mg, 3.27 mmol), tert-butyl (4-aminophenyl)carbamate (818 mg, 3.93 mmol), K2CO3 (678 mg, 4.91 mmol) in anhydrous DMF (5 mL) was stirred at room temperature for 5 h. After confirming the completion of the reaction, the mixture was quenched with water and then extracted with ethyl acetate (EA). The organic layer was combined, dried over MgSO4, filtered, and concentrated using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA = 4: 1) to give compound 50. Next, compound 50 (800 mg, 2.24 mmol) was placed in a round-bottom flask, and dissolved in methanol (10 mL). Then, 10 mL of LiOH aqueous solution (1.0 M) was added. The reaction mixture was refluxed at 60 °C for 3 h. The solvent was reduced by half by using a rotary evaporator, and then HCl 1.0 N was added to adjust the pH to 2-3. The resulting mixture was then extracted with EA, dried over MgSO4, filtered, and concentrated using a rotary evaporator to obtain a residue. The residue was then purified by silica column chromatography (DCM: MeOH = 4: 0.1) to give compound 51 in 54% yield.
Compound 51: 1H NMR (300 MHz, Methanol-d4) δH 7.96 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 8.1 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 6.57 – 6.54 (m, 2H), 4.36 (s, 2H), 1.48 (s, 9H).
4.1.34. tert-butyl (4-((4-((2-Aminophenyl)carbamoyl)benzyl)amino)phenyl)carbamate (52)
A mixture of 4-(((4-((tert-butoxycarbonyl)amino)phenyl)amino)methyl)benzoic acid 51 (300 mg, 0.88 mmol), o-phenylenediamine (107 mg, 1.06 mmol), EDC.HCl (203 mmol, 1.06 mmol), HOBt (178 mg, 1.32 mmol), N-methylmorpholine (242 µL, 2.2 mmol) in anhydrous DMF (4 mL) was stirred at room temperature for 5 h. The mixture was quenched with water and then extracted with EA. The organic layer was combined, dried over MgSO4, filtered, and concentrated using a rotary evaporator to obtain a residue. The resulting residue was then purified by silica column chromatography (n-hexane: EA = 2: 3) to give compound 52 in 86% yield.
Compound 52: 1H NMR (300 MHz, DMSO-d6) δH 9.58 (s, 1H), 8.77 (s, 1H), 7.92 – 7.89 (m, 2H), 7.47 – 7.44 (m, 2H), 7.17 – 7.07 (m, 3H), 6.99 – 6.94 (m, 1H), 6.78 – 6.76 (m, 1H), 6.61 – 6.56 (m, 1H), 6.49 – 6.46 (m, 2H), 6.07 (t, J = 6.2 Hz, 1H), 4.87 (s, 2H), 4.31 (d, J = 6.2 Hz, 2H), 1.43 (s, 9H).
4.1.35. N-(2-Aminophenyl)-4-(((4-aminophenyl)amino)methyl)benzamide (53)
Tert-butyl (4-((4-((2-Aminophenyl)carbamoyl)benzyl)amino)phenyl)carbamate 52 (327 mg, 0.76 mmol) was dissolved in anhydrous DCM (6 mL). About 2 mL of TFA was added dropwise under an ice bath. The resulting mixture was then stirred at room temperature for 2 h. After confirming the completion of the reaction, the solvent was removed using a rotary evaporator to obtain the crude product. It was triturated by the addition of ether and hexane to give compound 53 in 90% yield.
Compound 53: 1H NMR (300 MHz, DMSO-d6) δH 10.19 (s, 1H), 7.97 (d, J = 8.1 Hz, 2H), 7.50 (d, J = 8.1 Hz, 2H), 7.38 (d, J = 7.5 Hz, 1H), 7.25 – 7.15 (m, 4H), 7.04 (d, J = 8.7 Hz, 2H), 6.64 (d, J = 8.7 Hz, 2H), 4.39 (s, 2H).
4.1.36. N-(2-aminophenyl)-4-(((4-((6-chloro-5,8-dioxo-5,8-dihydroquinolin-7-yl)amino)phenyl)amino)methyl)benzamide (18A) and N-(2-aminophenyl)-4-(((4-((7-chloro-5,8-dioxo-5,8-dihydroquinolin-6-yl)amino)phenyl)amino)methyl)benzamide (18B)
A mixture of N-(2-aminophenyl)-4-(((4-aminophenyl)amino)methyl)benzamide 53 (200 mg, 0.60 mmol), 6,7-dichloroquinoline-5,8-dione (100 mg, 0.44 mmol), DIPEA (300 µL, 1.72 mmol) in MeOH (5 mL) was stirred at room temperature for 7 h. The mixture was concentrated under reduced pressure to obtain the crude product. The resulting crude product was purified by silica column chromatography (DCM: MeOH = 60: 1) to obtain the 7-, 6-substituted product 18A and 18B respectively.
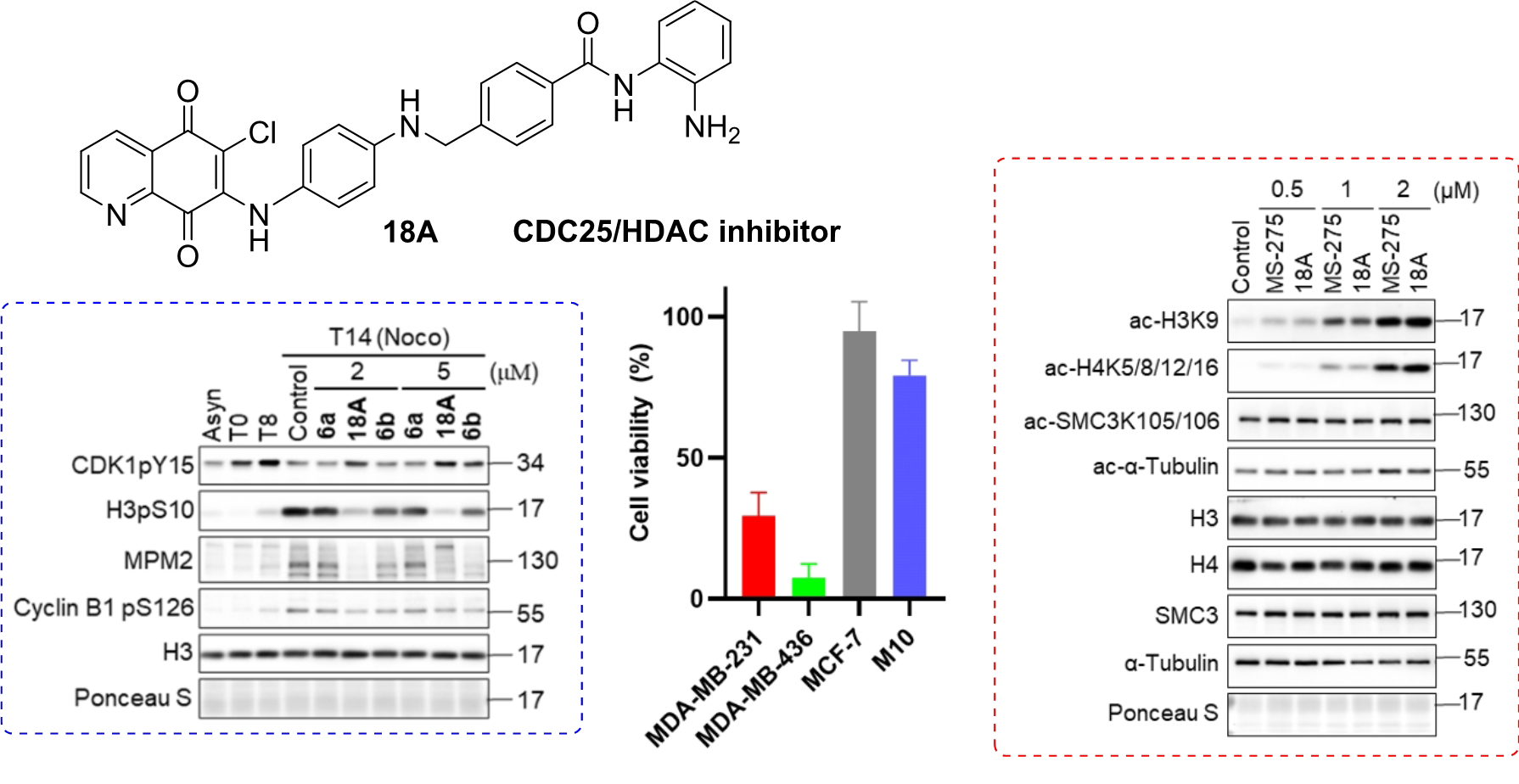
Compound 18A: m.p = 182-183 oC, purple solid, yield = 23%; 1H NMR (300 MHz, DMSO-d6) δH 9.60 (s, 1H), 9.26 (s, 1H), 8.92 (dd, J = 4.7, 1.7 Hz, 1H), 8.35 (dd, J = 7.9, 1.7 Hz, 1H), 7.94 (d, J = 8.1 Hz, 2H), 7.80 (dd, J = 8.1, 4.8 Hz, 1H), 7.49 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 6.6 Hz, 1H), 7.00 – 6.94 (m, 1H), 6.89 (d, J = 8.7 Hz, 2H), 6.77 (dd, J = 7.8, 1.2 Hz, 1H), 6.62 – 6.52 (m, 3H), 6.44 (t, J = 6.3 Hz, 1H), 4.88 (s, 2H), 4.37 (d, J = 5.7 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δC 178.5, 175.5, 165.2, 153.0, 146.4 (2C), 144.0 (2C), 143.0, 133.8, 133.1, 129.5, 128.4, 127.8, 127.4, 126.9, 126.6, 126.4, 125.9, 123.4, 116.2, 116.1, 111.5, 110.4, 46.3. HRMS (ESI) calcd for C29H23O3N5Cl [M+H]+ 524.1489; found 524.1483. HPLC purity = 97.3% (tr = 17.818 min.).
Compound 18B: m.p = 185-186 oC, purple solid, yield = 60%; 1H NMR (300 MHz, DMSO-d6) δH 9.61 (s, 1H), 9.16 (s, 1H), 8.96 (dd, J = 4.8, 1.7 Hz, 1H), 8.35 (dd, J = 7.8, 1.8 Hz, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.75 (dd, J = 8.1, 4.8 Hz, 1H), 7.49 (d, J = 8.1 Hz, 2H), 7.16 (d, J = 7.8 Hz, 1H), 6.99 – 6.94 (m, 1H), 6.91 – 6.87 (m, 2H), 6.77 (dd, J = 8.1, 1.2 Hz, 1H), 6.62 – 6.52 (m, 3H), 6.44 (t, J = 6.1 Hz, 1H), 4.88 (s, 2H), 4.37 (d, J = 6.0 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δC 180.1, 174.8, 165.2, 154.4, 148.0, 146.4, 144.0, 143.1, 142.8, 134.3, 133.1, 127.8, 127.4, 127.3, 126.9, 126.6, 126.4, 125.9, 123.4, 116.3, 116.1, 112.5, 111.5, 46.3. HRMS (ESI) calcd for C29H23O3N5Cl [M+H]+ 524.1489; found 524.1476. HPLC purity = 98.2% (tr = 17.259 min.).
4.2 Biology
4.2.1 Cell culture
Triple-negative breast cancer MDA-MB-231 and Pancreatic ductal adenocarcinoma PANC-1 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (12800-017; Gibco). Triple-negative breast cancer cells MDA-MB-436 were cultured in Dulbecco’s Modified Eagle Medium F12 (DMEM F12) medium (41300-039; Gibco). Human epidermal growth factor receptor-2 negative breast adenocarcinoma MCF-7 and gliobastoma U87MG cells were cultured in alpha modified Eagle's minimum essential medium (α-MEM) (11900-024; Gibco). Human leukemia cell lines KG-1, HL-60 and colorectal adenocarcinoma DLD-1 were cultured in Roswell Park Memorial Institute (RPMI) 1640 (31800-022; Gibco). Human leukemia cell lines MV4-11 and K-562 were cultured in Iscove’s modified Dulbecco’s medium (IMDM) (12200-036; Gibco), containing 1.5g/L and 3g/L sodium bicarbonate respectively. All cell lines were cultured in medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin-glutamine (PSG; 10378-016; Gibco) at 37 ◦C in a humidified atmosphere.
4.2.2. Chemicals and antibodies
Colchicine, Paclitaxel (Taxol) and Nocodazole were purchased from Sigma (C9754), Cytoskeleton, Inc. (TXD01) and Sigma (31430-18-9) respectively. Primary antibodies: anti-phospho-Ser/Thr-Pro-mitotic protein monoclonal 2 (MPM2) (05–368; Millipore); anti-cyclin B1 pS126 (ab55184; Abcam); anti-Cyclin-dependent kinase 1 pY15 (CDK1 pY15) (GTX1281550; GeneTex); anti-histone H3 pS10 (06–570; Millipore); anti-Histone 3 (ab1791; Abcam); anti-acetyl-Histone 3 lysine 9 (07e352; Millipore); anti-Histone 4 (ab10158; Abcam); antiacetyl-Histone 4 lysine 5/8/12/16 (06e866; Millipore); anti-SMC3 (A300-060A; Bethyl); anti-acetyl-SMC3 lysine 105/106 (MABE1073; Millipore); anti-a-tubulin (T5168; Sigma); anti-acetyl-a-Tubulin lysine 40 (T7451; Sigma); anti- KRAB domain-associated protein 1 (KAP1) pS824 (ab70369; Abcam); anti-Checkpoint kinase 2 (CHK2) pT68 (2661; Cell Signaling Technology); anti-CHK1 pS345 (2348; Cell Signaling Technology); anti-CHK1 (G-4) (sc-8408; Santa Cruz); anti-Replication Protein A2 (RPA2) pS33 (A300-246A; Bethyl); anti-RPA2 pS4/S8 (A300- 245A; Bethyl); anti-γH2AX (05–636; Millipore); anti-H2AX (2595; Cell Signaling Technology); anti-actin (MAB1501; Millipore); anti-Caspase-3 (NB100-56708; Novus); anti-Caspase-8 (9746; Cell Signaling); anti-Caspase-9 (9502; Cell Signaling); anti-induced myeloid leukemia cell differentiation protein (Mcl-1)(ab32087, Abcam); anti-B-cell leukemia/lymphoma 2(Bcl-2)(3498; Cell Signaling). Secondary antibodies: Horseradish peroxidase (HRP)-conjugated goat anti-mouse (115-035-003; Jackson ImmunoResearch Labs); anti-rabbit (111-035- 003; Jackson ImmunoResearch Labs) antibodies.
4.2.3. MTT assay
Cells were seeded in 96-well plates and treated with compounds for 48h, followed by incubation with 1 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma, M2128) at 37 ◦C for 2-3 h. Formazan crystals were then dissolved in dimethyl sulfoxide (DMSO). The number of surviving attached cells was determined by measuring the absorbance at 562 nm (PerkinElmer VICTOR3TM multilabel plate reader).
4.2.4. MTS assay
Cells were seeded in 96-well plates and treated with compounds for 48h. Surviving cells were determined by incubation with 0.2 mg/ml of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS; Abcam, ab223881), and absorbance at 490 nm was measured by using PerkinElmer VICTOR3TM multilabel plate reader.
4.2.5. Thymidine synchronization
Cells were incubated with 4 mM of thymidine (Sigma, T1895) for 20–24 h to enrich cells at the early-S phase. Cells were then washed with fresh medium twice and incubated with culture medium for recovery from thymidine block.
4.2.6. Flow cytometry
Cells were collected and fixed with 70% ice-cold ethanol. After washing with cold phosphate-buffered saline containing 1% FBS, cells were incubated with 0.05 mg/ml of propidium iodide (PI; Sigma, P4170) and 0.25 mg/ml ribonuclease A (RNase A; Sigma, R6513) at 37 ◦C for 30 min. DNA content was analyzed by using Becton Dickinson FACSCantoTMII Flow Cytometer, and cell-cycle profiles were plotted with the FlowJo software.
4.2.7. Western blotting analysis
Cells were lysed in Laemmli sample buffer (60 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate, and 10% glycerol). Proteins were separated by 12% SDS-polyacrylamide gel electrophoresis and transferred to the nitrocellulose membranes. After blocking with 5% skim milk, proteins were probed with specific primary antibodies and then HRP-conjugated species-specific secondary antibodies, followed by signal detection using enhanced chemiluminescence substrates (Bio-Rad). Images were obtained by using the iBright FL-1500 system.
{kind=link}
{kind=link}
{kind=link}
{kind=link}