Induced expression, purification, and detection of recombinant expression vector
Amplification and sequence analysis of the fnr and glnR genes
The complete genome DNA of L. plantarum WU14 was extracted using a bacterial genome extraction kit, and the results of the whole genome extraction are shown in Fig. 1A. Using the L. plantarum WU14 genome as a template, the fnr and glnR genes were amplified. The fnr gene was amplified using specific primers Fnr-F (Bam HⅠ) and Fnr-R (Xho Ⅰ), resulting in a size of 642 bp. Gel electrophoresis of the gel-recovered fragment is depicted in Fig. 1B. Similarly, the glnR gene was amplified using the same method, and the gel electrophoresis of the gel-recovered fragment is shown in Fig.1C.
Construction of fnr and glnR cloning vectors
The recovered fnr gene fragment was ligated into the pEASY-Blunt vector and transformed into E. coli TOP 10 competent cells. Eight transformed colonies were randomly selected and cultured overnight in LB liquid medium containing 600 μL 50 μg/mL Kanamycin. Using the M13-F/M13-R primers, bacterial liquid PCR verification was carried out, as depicted in Fig. 1D. The + lane served as a control using the empty vector as a template (band size of 150 bp), and the - lane used water as a negative control. The correct size for transformed colonies should exhibit the empty vector size plus the fnr gene fragment (band size of 800 bp). Bands in lanes 1-2 and 6-7 represented correctly sized transformed colonies. Colonies 1 and 6 were selected for plasmid extraction.
Using the same methodology, verification of the Blunt-glnR vector was conducted, yielding results as shown in Fig. 1E. The + lane served as a positive control (fragment size of 381 bp), and the - lane used water as a negative control. Consequently, bands in lanes 1-2 and 4-8 represented colonies with correct band sizes. Colonies 1 and 4 were chosen for plasmid extraction.
Plasmids from the selected four colonies were extracted and sent to the Genetic Sequencing Department at Shenggong Bio for sequencing. The sequencing verification and alignment results demonstrated the accuracy of the sequences.
After the correct Blunt fnr clone vector and pGEX-6p-1 empty vector digested with Bam HⅠ and Xho Ⅰ were sequenced, the correct bands were detected and recovered by 1% agarose gel electrophoresis. The fnr and pGEX-6p-1 fragments were ligated with T4 DNA ligase, transformed into the Transetta competent cells using heat shock, and coated on the LB flat plate with 50 μg/mL Amp for overnight culture. Then, 8 transformants were cultured in the liquid LB with 600 μL of 50μg/mL Amp and verified by PCR using Fnr-F and Fnr-R primers. As shown in Fig. 1F, the correct size of the bands 1-8 were all positive transformants, with the + lanes using the clonning vector as the template for the positive control (the strip size was 642 bp) and the - lanes used the water as the template for negative control; the No.1 clone was selected for subsequent induction expression experiment. The verification of the pGEX-6p-1-glnR recombinant expression vector was consistent with the above method. As shown in Fig. 1G, the correct strip size of 1-8 swimming lanes were all positive transformants, and the strip size should be empty plus glnR fragment (~500 bp) with the - lane used water as the template for the negative control, the K lane used the pGEX-6p-1 empty carrier as the template for negative control (~150 bp), and the + lane used the pGEX-6p-1-glnR vector as the template for positive control. The No.1 clone was selected for subsequent induction expression experiments.
Construction of fnr and glnR recombinant expression plasmids
Induction of recombinant fnr and glnR proteins expression
Using the bioinformatics software SnapGene, the fnr gene sequence was analyzed, revealing that the size of the Fnr protein is 25 kD. The GST protein tag in the pGEX-6p-1 vector is also 25 kD. Therefore, the induced protein size using this vector should be approximately 50 kD. The tag size in the pET30a vector can be disregarded. After culturing the bacterial strain, the cell lysate was collected through centrifugation. The supernatant was separated from the pellet (including the blank control) and subjected to SDS-PAGE analysis, as shown in Fig. 2A. When induced by the pET30a vector, the 25 kD band in the supernatant was weaker compared to the empty vector, while the concentration of the induced pellet band was significantly higher and formed aggregates. Thus, Fnr protein expression in the pET30a vector was associated with inclusion bodies. When induced by the pGEX-6p-1 vector, a visibly stronger band appeared at 50 kD in the supernatant compared to the empty vector supernatant. The induced pellet band was much higher than the empty vector pellet band, and aggregation was also observed. Hence, Fnr protein expression in the pGEX-6p-1 vector also involved inclusion bodies, with some expression present in the supernatant that could be purified and recovered.
By using the bioinformatics software SnapGene, the glnR gene sequence was analyzed, revealing that the theoretical size of the GlnR protein is 15 kD. Thus, the theoretical size of the recombinant protein is 40 kD. Induction of correctly verified bacterial strains was achieved using 0.1 mmol/L IPTG. Supernatant and pellet (including blank control) were analyzed through SDS-PAGE, as illustrated in Fig. 2B. At the 40 kD band size, the protein concentration in the induced supernatant was higher than in the empty vector, indicating inclusion body expression. However, soluble expression was also detected in the supernatant, which could be purified.
Purification and analysis of recombinant Fnr and GlnR proteins
The purified and recovered Fnr protein was subjected to SDS-PAGE analysis, as illustrated in Fig. 2C. Lanes 6-10 represent eluted target protein samples. In Lane 6, a distinct band around 50 kD is observed, corresponding to the expected size of the target protein. The purified protein displays minimal impurities, rendering it suitable for subsequent EMSA experiments. The purification method for GlnR protein is analogous to that of Fnr protein purification. After column penetration, a gradient elution with reduced glutathione was employed. The protein eluate was analyzed using SDS-PAGE, as depicted in Fig.2D. Lanes 5, 15, and 30 represent the original eluted protein samples, while lanes 5N, 15N, and 30N depict the protein samples after a 5-fold concentration using ultrafiltration. Notably, a single band is consistently observed around 40 kD in Figure 2D, aligning with the anticipated size of the target protein. The purified protein concentration is relatively low, but after concentration, it can be utilized for EMSA experiments.
EMSA verification of Fnr and Pnir promoters’ interaction
Promoter amplification
The PglnR promoter and the biotin-labeled PglnR promoter were amplified by the PglnR-F/PglnR-R and PglnR-FS/PglnR-RS specific primers using the L. plantarum WU14 genome as the template. The results were detected by 1% agarose gel electrophoresis, as shown in Fig. 3A. Since the promoter of the glnR gene was intercepted at 111 bp before the glnR gene, its theoretical size was 111 bp. The size of lane 1 was between 65-230 bp, and the possibility of primer dimer cloud be eliminated, which could be used as the target band for the next experiment. The Pnir promoter and the biotin-labeled Pnir promoter were amplified in the same way as the glnR gene promoter, and detected by 1% agarose gel electrophoresis, as shown in Fig. 3B. The first 400 bp of the nir gene was intercepted as the promoter of nir gene, so the theoretical size of Pnir was 400 bp. The 1-2 lanes in the figure were Pnir, and the migration speed of gel electrophoresis was not affected by the presence or absence of biotin markers. The band size was 400 bp, and the theoretical value was the same.
EMSA verification of Fnr and Pnir promoters’ interaction
In the purification process of the GST protein, the expression of E. coli transformed by the empty vector pGEX-6p-1 was induced. After the cell was broken, it was purified by a column using the recombinant fusion protein purification method. The eluant of the gradient elution was analyzed by SDS-PAGE, as shown in Fig. 3C. Since the theoretical size of GST was 25 kD, there was a single band of 25 kD in No. 1-2 lanes, and the concentration was high, indicating that the purification effect of GST protein was good, which could be preserved for later experiments.
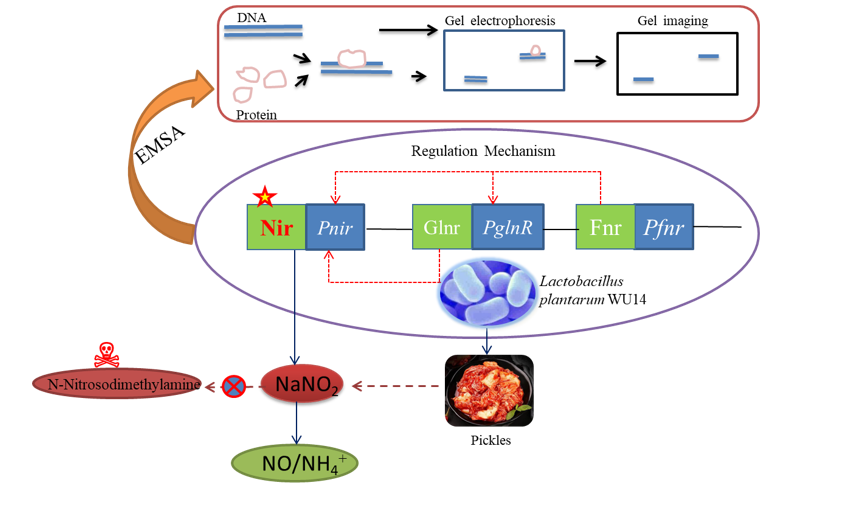
The EMSA could verify whether Fnrinteracted with the Pnir promoter in vitro. When the biotin probe DNA was added with 20 fmol, the self-luminescence of biotin showed visible bands. When the protein reacted with the probe DNA, the migration speed of the DNA protein complex on the active gel slowed down, and a hysteresis band appeared. This result showed that the GST protein did not specifically bind to the probe when the gradients of the recombinant protein and competitive DNA were set at the same time. As shown in Fig. 3D, there was only a probe band without a lagging binding band in the GST lane, indicating that the GST protein did not bind to the Pnir promoter. However, when the GST-Fnrprotein was added, a DNA protein complex retention band appeared, and the retention band increased with the increase of concentration, indicating that the GST-Fnrprotein could bind to the Pnir promoter in vitro. When the competitive DNA was added to the reaction system, the retention band become weaken and gradually disappeared with the increase in concentration. This might be because the concentration of the competitive DNA was much higher than the probe DNA. When it was bound to the protein, only a few or no probe DNA was bound to GST-Fnr, and the complex lagging band weakened or disappeared. These results showed that the Fnr protein could bind to the Pnir promoter in vitro and regulate the expression of the nir gene.
EMSA verification of Fnr and PglnR promoters’ interaction
In this study, the interaction between Fnrand PglnR promoters was explored in vitro to prove that Fnr could regulate Nirby regulating glnR indirectly. Moreover, whether Fnrcould bind to the PglnR promoter in vitro was verified through the EMSA experiment. As shown in Fig. 3E, no binding hysteresis band was found in the GST lane, indicating that the GST protein did not bind to the PglnR promoter; however, the retention bands were obvious in the lane added with the GST-Fnrprotein, indicating that the Fnrprotein interacted with the PglnR promoter in vitro. After adding the competitive DNA, the retention bands were weakened significantly and gradually disappeared with the increase of concentration, indicating that the Fnr protein bound to the PglnR promoter. The EMSA experiment results showed that Fnrhad a regulatory effect on the transcription of glnR. The combined research the above proved that Fnrcould indirectly regulate the nir gene by regulating glnR.
Growth curve under different culture conditions and test of ability to degrade NaNO2
Growth curve of L. plantarum WU14 under O, A, NO, and NA condition (0.1% NaNO2)
The growth curve of L. plantarum WU14 cultured under O, NO, A, and NA conditions is shown in Fig. 4A. During 4-12 h culture, the strain was in a logarithmic growth period and became stable at 12 h-24 h. Only in the anaerobic culture, the strain was still growing slowly in the 12-24 h culture, which was better than that in the aerobic culture. The growth of the strain and the maximum concentration of the strain at the later stage were inhibited by 0.1% NaNO2.
Growth curve of L. plantarum WU14 under NaNO2 concentration gradient stress
The growth curve of L. plantarum WU14 under concentration gradient NaNO2 stress aerobic culture is shown in Fig. 4B. With the increase of NaNO2 concentration, the inhibition degree of cell growth increased, and the maximum cell concentration decreased with the increase of NaNO2 concentration. When the NaNO2 content was 2%, the cell growth increased after 12 hours of culture, and the maximum cell concentration was low with OD600 less than 1, indicating that the higher the concentration of NaNO2, the more obvious the inhibition degree of the cell.
The growth curve of L. plantarum WU14 anaerobic culture under NaNO2 concentration gradient stress is shown in Fig. 4D. With the increase of NaNO2 concentration, the inhibition degree of cell growth increased. Compared with Figures 4B and 4D, it was found that the maximum cell concentration value was greater under anaerobic conditions than under aerobic conditions, suggesting that L. plantarum WU14 has a stronger growth ability under anaerobic conditions than aerobic conditions.
Test of ability to degrade NaNO2
The change in NaNO2 content in the medium with time under the stress of different NaNO2 concentrationsis shown in Fig. 4C. With the increase of NaNO2 concentration, the amount of NaNO2 degraded by the strain decreased, indicating that its degradation ability was inhibited. When the content of NaNO2 was lower than 0.3%, the final degradation rate of NaNO2 exceeded 70%.
The change in NaNO2 content in the medium with time under the stress of different NaNO2 concentrations in anaerobic culture is shown in Fig. 4E. The change in the degradation ability of the strain was consistent with that under aerobic conditions; however, the degradation ability of the strain under anaerobic conditions was higher than that under aerobic conditions.
Verification of relative expression of fnr and nir by qRT-PCR
EMSA experiments confirmed that both Fnr and GlnR proteins are capable of binding to the Pnir promoter, regulating Nir activity. Fnr protein responds to changes in oxygen concentration, adapting cellular respiration to optimize cell growth under varying oxygen levels. GlnR, also known as a nitrogen metabolism regulatory protein, is sensitive to nitrite concentrations in the environment. Therefore, this experiment aimed to assess the expression of the fnr and nir genes by altering the bacterial growth environment.
The expression amount of fnr from L. plantarum WU14 with 16S rDNA as the internal reference gene under different culture conditions is shown in Fig. 5. The analysis of A and C histograms showed that the relative expression of fnr was relatively uniform within 6-12 h. The expression of fnr under anaerobic conditions was upregulated compared with that under aerobic conditions, and the gene expression under NaNO2 stress was also slightly upregulated. However, a relatively uniform conclusion could not be drawn by the analysis of other conditions and time points. Therefore, it was speculated that the change in the culture conditions alone might not have much impact on gene expression.
The expression of nir under different culture conditions was compared with that of internal reference 16S rDNA, as shown in Fig. 6. The histogram analysis indicated that there was no significant difference between the relative expression of nir and fnr. The expression of nir in the anaerobic culture was upregulated compared with the aerobic culture within 6-12 h, and the gene expression was also slightly upregulated under NaNO2 stress. Under anaerobic conditions, the expression of fnr and nir was higher than that under aerobic conditions within 6-12 h of culture, and the expression of these two genes increased with the addition of NaNO2 to the aerobic culture. However, under the conditions of anaerobic and NaNO2 stress, this phenomenon disappeared. It might be due to the complex regulation network of the cell, which affected other regulatory genes and had an impact on the results, leading to a low repeatability of the experimental results.
{kind=link}