3.1 Principle of the proposed ECL biosensor
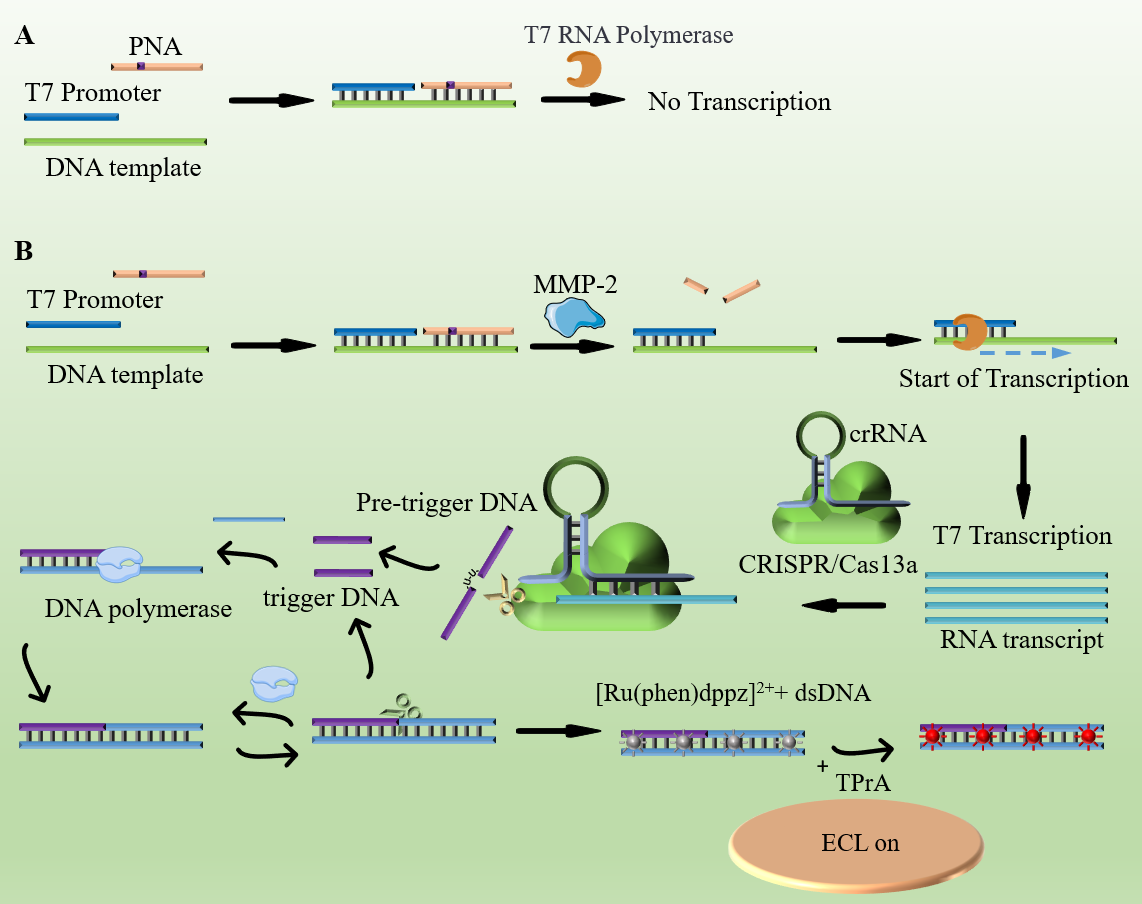
Scheme 1 provides an illustration of the platform and its operational principles. In this context, MMP-2 was chosen as the model target for the proof-of-concept. The Cas13a enzyme employed in this investigation is derived from Leptotrichia buccalis (LbuCas13a). The crRNA consists of a 30 nucleotide repeat region facilitating interaction with Cas13a, and a 20 nucleotide programmable guide region, often referred to as the spacer, designed for the recognition of target RNA. Notably, the trans cleavage activity of LbuCas13a exhibits a preference for cleaving the ribose-phosphodiester bond flanked by uracil. To leverage this preference, a Pre-trigger DNA was designed, incorporating two uracil ribonucleotides (rU). The 5′ end of this Pre-trigger DNA comprises 16 complementary bases with the amplification template and five random bases at the opposite end. The amplification template features a central nicking endonuclease (NEase) cleavage site and two separate repeat sequences, both of which are complementary to the 5′ end of the Pre-trigger DNA. Additionally, both the Pre-trigger DNA and the amplification template were equipped with a C3 spacer at their 3′-end to prevent target-independent polymerase amplification. A specific PNA sequence was designed, containing a peptide sequence (Gly-Pro-Leu-Gly-Val-Arg-Gly) that can be precisely cleaved by MMP-2. It is essential to note that PNA is a synthetic nucleic acid analog characterized by a peptide backbone rather than a sugar-phosphate backbone.
In the context of the assay reaction, the process unfolds as follows: Initially, the PNA/T7 promoter/DNA template complex, featuring a double-stranded DNA template connected by PNA and T7 promoter, which encompasses a peptide bond cleavable by MMP-2. In the absence of MMP-2 within the system, the PNA/DNA template duplex inhibits the T7 promoter amplification reaction (as depicted in Scheme 1A).
However, with the introduction of MMP-2 into the system (as presented in Scheme 1B), the cleavage of the peptide bond is triggered, leading to the release of the single-stranded DNA template/T7 promoter duplex. This released duplex assumes the role of a substrate for T7 promoter-aided transcription amplification. Subsequent transcription amplification generates a substantial quantity of single-stranded RNA transcripts, encompassing the target sequence that is identifiable and bindable by gRNA. Following the binding of gRNA to the transcript, it activates the nuclease activity of CRISPR/Cas13a, thereby enabling the cleavage of any single-stranded RNA molecules.
The assembled Cas13a/crRNA system is designed to specifically recognize and cleave target RNA sequences. Upon recognition, Cas13a's two conserved higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domains reposition to form a new RNase active site, activating its trans-cleavage activity. This leads to the cleavage of the Pre-trigger DNA into two fragments. The 5′ fragment of the cleaved Pre-trigger DNA can hybridize with an amplification template, initiating a polymerization reaction in the presence of T4 polynucleotide kinase (PNK) and Vent DNA polymerase. The newly synthesized double-stranded DNA is then recognized and nicked by nicking endonuclease (NEase). This process involves a continuous cycle of nicking, strand extension, and displacement, resulting in the generation of substantial amounts of double-stranded DNA products. Notably, the trans-cleavage activity of Cas13a remains inactive without the presence of the specific portion of the Pre-trigger DNA. If the Pre-trigger DNA is not cleaved, it cannot be extended by DNA polymerase, thereby failing to initiate the downstream exponential amplification reaction (EXPAR). This mechanism ensures that only in the presence of the target RNA does the system proceed, thereby providing high specificity and sensitivity in detecting the target sequence. This feature is critical for applications requiring precise molecular detection, such as the developed ECL biosensing platform for MMP-2 detection.
Subsequently, the complex [Ru(phen)2dppz]2 + is incorporated into the Cas13a-enhanced EXPAR (CAS-EXPAR) system. In its free state, this complex fails to emit luminescence due to nitrogen protonation in aqueous environments. However, when it binds to the double-stranded DNA products produced by EXPAR, there is a significant increase in luminescence. This boost in signal is linked to the planar phenazine ligand of [Ru(phen)2dppz]2+ engaging with the base pairs in the major groove of the DNA. This interaction shelters the phenazine's nitrogen atoms, promoting a state conducive to luminescence.
Following this, the [Ru(phen)2dppz]2+ supplemented amplification system and excess TPrA are introduced into the biosensor platform. The electrochemiluminescence (ECL) signal, which serves as an indicator of MMP-2 activity, is detected through a photomultiplier tube (PMT, Hamamatsu R928 PMT). The reaction mechanism is as follows:
[Ru(phen)2dppz]2+/DNA-e− →[Ru(phen)2dppz]3++H+
TPrA-e− → TPrA∙+ → TPrA∙ + H+
[Ru(phen)2dppz]3+-DNA + TPrA∙ → [Ru(phen)2dppz]2+∗-DNA + products
[Ru(phen)2dppz]2+∗-DNA → [Ru(phen)2dppz]2+-DNA + hν
3.2 Feasibility study
The CEISPR/Cas13a-based amplification system was subjected to thorough examination with regards to its electrochemiluminescence (ECL) performance. The results revealed a substantial enhancement in the ECL signal when the system was employed for detecting as low as 1 pM of MMP-2 (curve c). In stark contrast, the ECL signals observed were notably diminished in the absence of MMP-2 (curve a) or in the presence of a substantially lower concentration of MMP-2 (100 aM, curve b), as illustrated in Fig. 1. These findings underscore the crucial role of the target MMP-2 in initiating the biosensor's activation.
In response to the need for characterization of the EXPAR amplification and the cutting performance of Cas13a, we have conducted the necessary experiments. The results are detailed in Figures S1 and S2. Figure S1 presents the electrophoresis identification of the T7 RNA polymerase transcription-mediated amplification process, confirming the feasibility of the EXPAR amplification. Additionally, Figure S2 illustrates the cutting performance of Cas13a, demonstrating its efficiency in the process. These characterizations verify the reliability and effectiveness of the amplification and cleavage mechanisms used in our study.
3.3 Optimization of experimental parameters
In our pursuit of enhancing the performance of the biosensor, we meticulously conducted a comprehensive optimization process, focusing on three pivotal experimental parameters. Employing ECL, we conducted an in-depth examination of the biosensor's current response under various conditions and compared it to a control group (as illustrated in Fig. 2). The parameters that underwent meticulous optimization included:
Optimizing T7 RNA Polymerase Transcription Duration: As revealed in Fig. 2A, the ECL signal achieved stability after 30 minutes. This observation was the result of a progressive sequence from curve a to curve g, with time intervals set at 5, 10, 20, 30, 40, 50, and 60 minutes. This analysis pinpointed the ideal duration for T7 RNA polymerase transcription, which was determined to be 30 minutes.
Fine-Tuning CRISPR/Cas13a Reaction Time: As portrayed in Fig. 2B, the ECL signal exhibited a gradual decline from 5 minutes to 30 minutes, corresponding to time intervals of 5, 10, 15, 20, 25, and 30 minutes. Ultimately, stability was reached after 25 minutes, underscoring the optimal 25-minute CRISPR/Cas13a reaction time that consistently yielded the most favorable results.
Optimizing Cas13a/crRNA Concentration: The concentration of Cas13a/crRNA holds direct sway over its intrinsic cis-cleavage efficiency, consequently impacting the subsequent isothermal amplification efficiency. We introduced differing concentrations of Cas13a/crRNA (2.5 nM, 5 nM, 10 nM, and 20 nM) into the sensing system in a 1:1 ratio and subjected them to ECL analysis. Figure 2C clearly indicates that the sensor treated with 10 nM Cas13a/crRNA exhibited the highest signal. However, once the concentration of Cas13a/crRNA was elevated to 20 nM, the ECL signal ceased to increase, signifying that an excess of Cas13a/crRNA failed to further enhance the ECL signal. Consequently, for the ensuing research, we opted for a 10 nM concentration of Cas13a/crRNA.
This meticulous series of optimization steps not only served to amplify the biosensor's performance but also significantly bolstered its precision in the detection and quantification of MMP-2.
3.4 Detection of MMP-2 with the biosensor
To quantify MMP-2 biomarkers accurately, we established a robust correlation between the peak intensities of the Electrochemiluminescence (ECL) signal, denoted as ERu, as depicted in Fig. 3A and B (Time vs. ECL intensity and Potential vs. ECL intensity respectively), and the MMP-2 concentration, labeled as CMMP−2 (Fig. 3C). The observed linear correlation between ERu and CMMP−2 spans concentrations ranging from 10 aM to 100 pM. From the data, we formulated a regression equation: ERu (a. u.) = 0.091 × lgCMMP−2 + 0.040 (R² = 0.9976, n = 5). The linear regression equation in Fig. 3C is established using data from both Fig. 3A and Fig. 3B. The signal values in Figs. 3A and 3B are consistent, with the only difference being that the horizontal axis in Fig. 3A corresponds to Time, while in Fig. 3B it corresponds to Potential. This equation not only facilitates precise logarithmic quantification of MMP-2 in biological samples but also confirms the biosensor’s high precision and reliability. The detection limit (LOD), a critical metric of the biosensor’s sensitivity, was determined using the 3σ method, resulting in an LOD of 12.8 aM. This LOD notably outperforms those of many existing methodologies, highlighting our biosensor’s superior sensitivity and its ability to detect extremely low concentrations of MMP-2.
To provide a broader context, Table 1 contrasts our newly developed methodology with existing MMP-2 quantification techniques. The comparative analysis demonstrates that our approach provides comparable sensitivity and a linear detection range similar to current methods but with a slightly more advantageous detection limit. Such findings suggest that our biosensor not only meets but potentially exceeds current MMP-2 analysis standards, making it a promising alternative for anti-aging research applications, especially in diagnostic and monitoring scenarios.
Table 1
Comparison of different methods for MMP-2 assay.
| Method | LOD | Linear Range | Reference |
| Fluorescent Nanoprobe | 32 pM | 0.1–20 nM | [55] |
| Silicon Nanowire-Based Biosensor | 0.1 pM | 100 fM-10 nM | [5] |
| CRISPR Cas13a based biosensor | 62.05 fM | 150–2000 fM | [33] |
| Bipedal walking robot | 12.8 aM | 0.01–100 pM | This work |
3.5 Specificity and reproducibility of the strategy
Specificity is crucial in developing enzymatic methodologies, especially when employing Peptide Nucleic Acid (PNA) as a substrate to detect matrix metalloproteinase 2 (MMP-2). Ensuring that MMP-2 selectively cleaves the PNA substrate is essential for reducing unintended off-target interactions and increasing the accuracy of the detection method. To ascertain the specificity of this technique, we identified several proteins unlikely to interfere with PNA, including Esterase, Matrix Metalloproteinase 1 (MMP1), Thrombin, Bovine Serum Albumin (BSA), Alpha-fetoprotein (AFP), and Carcinoembryonic Antigen (CEA), used as reference proteins (illustrated in Fig. 4A). We measured their electrochemiluminescent (ECL) responses using the CRISPR/Cas13a amplification method. As demonstrated in Fig. 4A, the ECL readings for these non-target proteins were compared against those for MMP-2 (concentration of 500 fM). The findings clearly showed that MMP-2's ECL signal underwent the most notable change compared to the baseline, whereas the signals from the other proteins remained largely consistent with the blank controls. In conclusion, the effectiveness of using PNA as a substrate for MMP-2 detection heavily depends on the specificity of the interaction. By carefully selecting reference proteins and fine-tuning the PNA sequence to specifically recognize a target site within MMP-2, we can significantly enhance the method's specificity and minimize the likelihood of off-target effects. The electrochemiluminescent biosensor developed via the CRISPR/Cas13a amplification method showcases exceptional selectivity, positioning it as an effective tool for various applications in biotechnology and medical diagnostics.
Subsequently, we conducted an assessment of the stability of the biosensing platform. Figure 4B provides a detailed representation of the detection stability within this Electrochemiluminescence (ECL) detection platform. This figure depicts the detection signal generated by the biosensing platform when detecting 1 pM MMP-2 as the target analyte. Remarkably, as depicted in Fig. 4B, the detection signal for 1 pM MMP-2 remains consistently stable, even after subjecting it to ten consecutive scans, each spaced an hour apart (RSD = 1.34%). There is no substantial decline in the signal's intensity. These observations solidify the remarkable stability of our sensing platform.
Furthermore, we carried out an evaluation of the biosensor's shelf life. The biosensors, containing DNA/[Ru(phen)2dppz]2+ complexes, were stored in a sealed container at a constant temperature of 4°C, shielded from light, following the detection of 1 pM MMP-2. After each test, the ECL signal of the biosensor was measured, and the relative standard deviation (RSD) of the signal over the course of 14 days was calculated (as shown in Fig. 4C). Notably, the functionality and sensitivity of the CRISPR/Cas13a-based biosensor remained consistent throughout the entire testing period, exhibiting no noticeable performance degradation. The RSD value of the ECL signal was a mere 0.88%, underscoring the biosensor's high stability and repeatability.
3.6 Recovery studies of the biosensor in real samples
Biosensors based on electrochemiluminescence, engineered through the CRISPR/Cas13a isothermal amplification method, display an impressive blend of heightened sensitivity and precision. These qualities render them highly effective for detecting MMP-2 activity within complex biological matrices. Owing to the prevalent surge in MMP-2 activity across a variety of human cancers and tissues, targeting MMP-2 has become a focal point in the realms of cancer diagnostics and treatment. Faced with the challenge of scarce aging-related cell samples, this study utilized supernatants from HepG2 and LO2 cell cultures to evaluate the biosensor's efficacy. As depicted in Fig. 5, the examination of these supernatants indicated a negligible variation in electrogenerated chemiluminescence (ECL) signals among the LO2 cell samples. In contrast, the HepG2 cell samples exhibited a significant surge in ECL signals, marking a 6.73-fold increase relative to the LO2 samples. This significant increase is attributed to the heightened expression of MMP-2 in cancerous cells.
To ensure the specificity of our electrochemiluminescence sensor, we performed a targeted pre-treatment experiment involving HepG2 cells and a known MMP-2 inhibitor, ARP 100. This intervention significantly reduced the ECL signal change to levels comparable with those of the control group, nearly zero. Such a result strongly indicates that the ECL response is directly attributable to MMP-2's specific activity on the PNA substrate. This validation highlights the exceptional potential of electrochemiluminescence sensors utilizing the bipedal walker isothermal amplification method for detecting MMP-2 activity in clinical settings. These findings are crucial as they confirm the sensor's high specificity and sensitivity, demonstrating its significant advantages for practical applications in the medical field, particularly in cancer diagnostics and therapy development. The technology’s precise detection capabilities enable it to identify MMP-2 activity accurately, making it a valuable tool for developing targeted treatment strategies and improving patient outcomes in oncology.
Subsequently, we measured the content and recovery rate of matrix metalloproteinase-2 (MMP-2) by correlating it with the spectrum of electrochemiluminescence (ECL) signals derived from LO2 cell culture supernatants. These signal levels fluctuated between 99.8% and 105.0%, as outlined in Table 2. This significant finding highlights the biosensor's outstanding ability to withstand external disturbances, making it an excellent candidate for reducing CRISPR/Cas13a activity, especially under complex conditions. The findings from these tests conclusively demonstrate the superior analytical capabilities of our approach in detecting MMP-2.
The ECL signal levels we obtained play a crucial role in deciphering both the concentration and recovery of MMP-2, offering a reliable method for its assessment and quantification across various sample types. The wide range of recovery rates demonstrates the robust stability of our biosensors, even in the face of potential disruptions caused by background noise, contaminants, or other biological elements prevalent in real-world specimens. This resistance to interference is particularly valuable in practical scenarios, where dealing with complex biological matrices demands high precision and specificity in detection.
Table 2
Recovery results for the assay of MMP-2 in cell culture supernatants of LO2.
| Sample number | Added (aM) | Found (fM) | Recovery (%) | RSD (%, n = 3) |
| 1 | 0 | 2.6 | | |
| 3 | 100 | 103.2 | 100.6 | 3.73 |
| 4 | 1000 | 1052.6 | 105.0 | 4.65 |
| 5 | 10000 | 9979.8 | 99.8 | 4.62 |
| 6 | 100000 | 100159.8 | 100.2 | 3.63 |
{kind=link}