3.1 Synthesis of Oxidized Chondroitin Sulfate (OCS)
OCS was synthesized by an oxidation reaction using NaIO4. First, 1.0 g of chondroitin sulfate (CS) was dissolved in 20 mL distilled water. Then, 700 mg of NaIO4 was dissolved in 20 mL distilled water. After dissolving completely, two solutions were mixed and stirred for 6 h in the dark at room temperature. Subsequently, 1 mL of ethylene glycol was added into the reaction system to terminate the oxidation reaction for 1 h. And then, the solution was dialyzed with distilled water for 3 days and freeze-dried, resulting in a white solid sample. OCS was stored at room temperature and protected away from light.
3.2 Fabrication of pH/Ultrasound Dual-Responsive Hydrogel
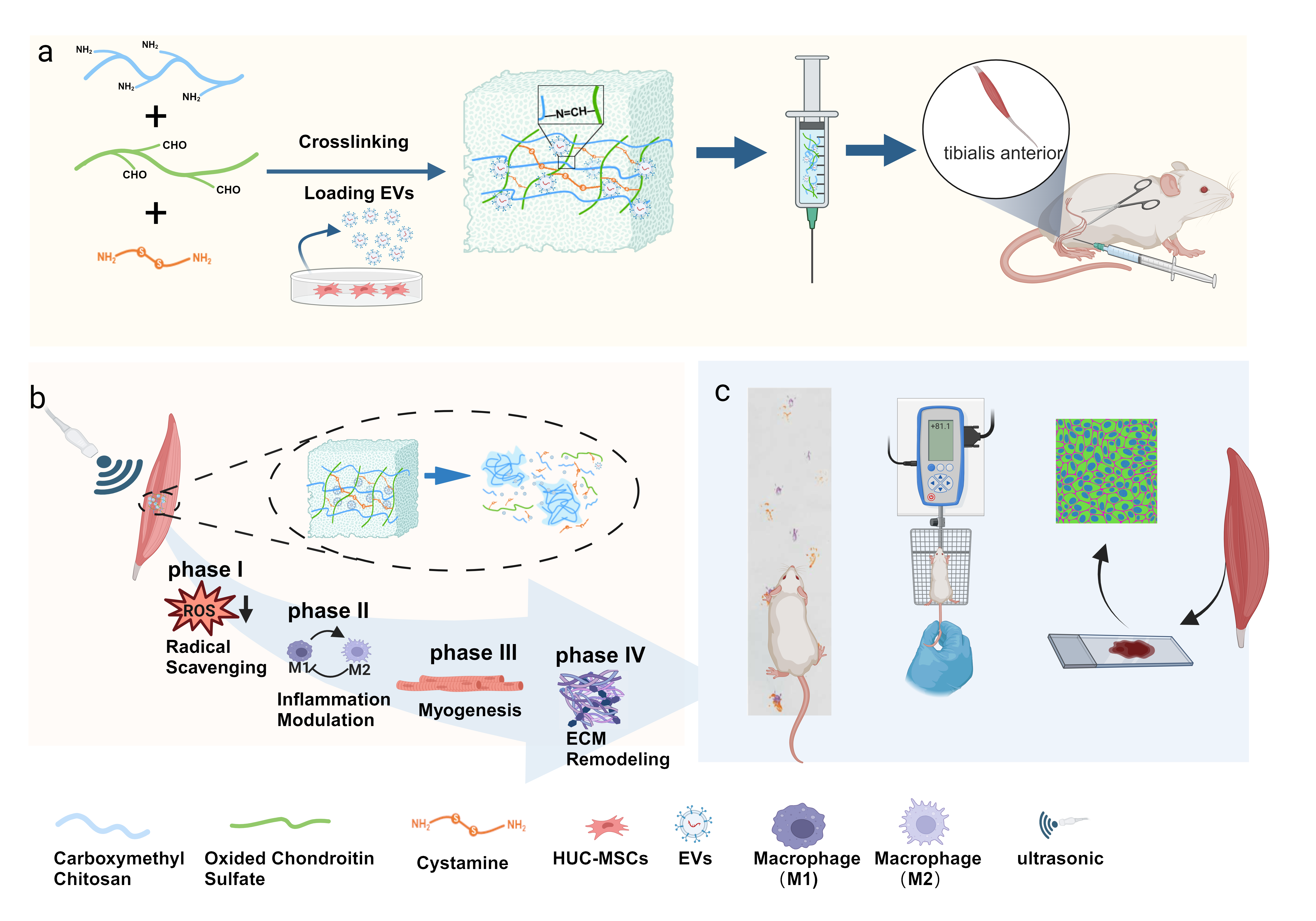
The pH/ultrasound dual-responsive hydrogel was prepared based on the principle of the Schiff base reaction. In brief, carboxymethyl chitosan (CMCS, 1.0 g) was dissolved in 50 mL of deionized water with stirring to completely dissolved. OCS (500 mg) was dissolved in 5 mL of deionized water to obtain 100 mg/mL of OCS. Cystamine dihydrochloride was dissolved in deionized water to 1.0 mol/L. And then, CMCS, OCS and cystamine dihydrochloride solutions were mixed at a volume ratio of 8:1:1 at room temperature with slight stirring. The pH/ultrasound dual-responsive hydrogel was gradually solidified and fabricated.
3.3 Fourier Transform Infrared Spectra (FTIR) Measurement
FTIR data were recorded using a Bruker Equinox 55 spectrometer at frequencies ranging from 400 cm− 1 to 4000 cm− 1 and resolution of 0.5 cm–1. Samples were powdered and mixed with dried KBr powder and pressed into pellet form.
3.4 Morphology of Hydrogels
The morphologies of hydrogels were analyzed by scanning electron microscopy (SEM, Zeiss microscope). Samples were washed for three time with deionized water and frozen in -20 ℃. Then the samples were freeze-dried to obtain anhydrous samples. Both hydrogels before and after ultrasound treatment were observed by SEM.
3.5 Rheology Test of Hydrogels
The hydrogels were performed on HAKKE rheometer to study the rheological property. The viscoelastic properties of the hydrogels were measured by performing strain sweep experiments in the oscillation mode. The frequency was set at 1 Hz, and the storage modulus (G′) and loss modulus (G″) values were recorded by sweeping tests changing the strain from 0.1–1000%. The hydrogels were divided into two groups, one untreated and the other sonicated by ultrasound diagnostic equipment (1.0 mW/cm2, 10 min).
3.6 Swelling Behavior Studies of Hydrogels
Anhydrous hydrogels after freeze-drying were weighed (WD), and then stored in PBS buffer to allow water uptake. The swollen hydrogels were extracted and weighed (WS) after wiping to remove excessive water at different time points, and the WS/WD ratio was calculated. The experiment was measured continuously for 10 h to fully analyze the swelling behavior of the hydrogels.
3.7 Biodegradation Behavior of Hydrogel
Anhydrous hydrogels after freeze-drying were weighed (W0) and immersed in PBS buffer (pH = 7.4) with 0.4 mg/mL of lysozyme (1 × 105 U·mg) at 37 ℃. The hydrogels were taken out and weighed (Ws) at different times. The experiment was measured continuously for 6 d to fully analyze the biodegradation behavior of the hydrogels. The degradation rates were calculated using the following formula:
The degradation rate = (1 – Ws/W0) × 100%
3.8 In vitro Drug Release
The in vitro drug release behavior of the pH/ultrasound dual-responsive hydrogel were investigated using methylene blue (MB) as the model drug. First, a range of concentrations of MB from 0 to 25 µg/mL were prepared. And the absorbance was measured by UV-visible spectroscopy (INSEA, China). The peak absorbance at 465 nm were recorded to fit the calibration curve of MP. And then, 20 µL of MB solution (1.0 mg/mL) was added into the 980 µL of the uncross-linked hydrogel solution to prepare MB-loaded hydrogel (20 µg MB per 1.0 mL hydrogel). The hydrogel of free releasing was set as control. The MB-loaded hydrogels were immersed in 5.0 mL PBS buffer (pH 7.4) to study release behavior. And hydrogels of ultrasound treatment, pH treatment (immersing into pH 6.0 PBS buffer), ultrasound + pH treatment were set as experimental group. The ultrasound treatment was set at 60th, 120th, 180th, and 300th min, and each ultrasound treatment lasted 10 min. The absorbance of MB in the were measured by UV-visible spectroscopy and the released percentages were calculated by the fitted calibration curve.
3.9 Cell Acquisition and Culture
As previously described30, 38, human umbilical cord mesenchymal stem cells (Cyagen, HUXUC-01001, China) were purchased from Cyagen Biotechnology Company. The HUC-MSCs were cultured in MesenCult™ MSC Basal Medium (Stemcell Technologies, RC200133, China). The multilineage differentiation potential of HUC-MSCs was verified by inducing osteogenic, chondrogenic, and adipogenic differentiation using differentiation media. Surface markers of HUC-MSCs (CD105, CD73, CD90, CD45, CD34, HLA-DR) were detected using flow cytometry.C2C12 myoblast cell line was purchased from Procella (Procella, CL-0044, China). The cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 IU/ml penicillin, and 100 µg/mL streptomycin. When the myoblasts reached 70%-80% confluence, they were induced to differentiate into myotubes using DMEM supplemented with 2% horse serum, 100 IU/mL penicillin, and 100 µg/mL streptomycin. RAW cells (RAW 264.7, a murine-derived macrophage cell line) were obtained from the cell bank of the Chinese Academy of Sciences Typical Culture Preservation Committee.
3.10 EVs Isolation and Characterization
The supernatant from HUC-MSCs culture was collected for EVs isolation using ultracentrifugation. The specific steps were as follows: The collected cell supernatant was centrifuged at 300 g, 4°C for 10 minutes to remove cell debris. The supernatant was then centrifuged at 2000 g, 4°C for 10 minutes to remove dead cell debris. Subsequently, the supernatant was centrifuged at 10,000 g, 4°C for 30 minutes to remove cellular debris. The resulting supernatant was filtered through a 0.22 µm filter and transferred to ultracentrifuge tubes. Ultracentrifugation was performed at 100,000 g (Beckman Coulter, USA) for 70 minutes at 4°C. The pellet obtained after centrifugation was resuspended in 1ml PBS52. Next, Nanosight NS300 system (Malvern, UK) was used for nanoparticle tracking analysis (NTA) to analyze the size distribution and particle concentration of the vesicles. Western blotting was then performed to detect the surface markers CD63, CD81, and CD9 of the EVs (Abcam, USA).
3.11 PKH-26 Staining and Cellular Uptake Assay
The PKH-26 staining kit (Sigma-Aldrich, D0030, China) was employed to label the EVs. The brief procedure, as provided by the manufacturer, is outlined below: The previously isolated EVs were diluted with Dilution C solution, followed by the addition of 4 µL of PKH-26 dye solution. The mixture was then incubated at room temperature in the dark for 5 minutes. Subsequently, the staining reaction was terminated by adding 500 µL of 1% BSA solution. The labeled EVs were pelleted by centrifugation at 1000000 g for 70 minutes at 4°C, followed by resuspension in 200 µL of cold PBS. C2C12 cells were co-cultured with the labeled EVs for 24 hours. The cells were then fixed with 4% paraformaldehyde, stained with DAPI for 5 minutes to label the cell nuclei, and observed under a fluorescence microscope to visualize the stained cells.
3.12 MiRNA Sequencing Analysis
Total RNA was extracted from the tissue using TRIzol® Reagent according the manufacturer’s instructions. Then RNA quality was determined by 5300 Bioanalyser (Agilent) and quantified using the ND-2000 (NanoDrop Technologies). Only high-quality RNA sample was used to construct sequencing library. RNA purification, reverse transcription, library construction and sequencing were performed at Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (Shanghai, China) according to the manufacturer’s instructions (Illumina, San Diego, CA).
3.13 Proliferation Experiment of C2C12 Cells
The proliferation assay of C2C12 cells was conducted using the CCK-8 cell counting kit (CCK-8, Dojindo, Japan) to assess the proliferation of cells stimulated by EVs. In brief, 5000 cells were seeded in a 96-well plate and cultured for 24 hours. Subsequently, 10 µL of CCK-8 solution was added to each well, followed by co-incubation with the cells for 3 hours. Cell proliferation was determined by measuring the absorbance at 450 nm using an enzyme-linked immunosorbent assay plate reader. The data were expressed as mean ± standard deviation (SD) from three independent replicates.
3.14 Immunofluorescence Experiment of C2C12 Cells
The immunofluorescence experiment of C2C12 cells (MYOG, DAPI staining) proceeded as follows: After differentiation in high-glucose DMEM medium containing 10% FBS for 3 days, cells in logarithmic growth phase were seeded onto coverslips to form a monolayer. Subsequently, cells were cultured in high-glucose DMEM medium supplemented with 2% horse serum for 7 days. Following this, cells were fixed with 4% paraformaldehyde at room temperature for 30 minutes, washed three times with 1× PBS for 5 minutes each. Non-specific sites were then blocked with 1% bovine serum albumin (BSA) at room temperature for 30 minutes. Each coverslip was incubated overnight at 4°C with mouse monoclonal anti-myogenin primary antibody (1:200 dilution in 1% BSA/PBS) (Beyotime, AF7542, China), followed by three washes with 1× PBS for 5 minutes each. Subsequently, coverslips were incubated with FITC-conjugated secondary antibody (anti-rabbit or anti-mouse) (1:500 dilution in 1% BSA/PBS) (Beyotime, A0562, China) at room temperature in the dark for more than 1 hour, followed by three washes with 1× PBS for 5 minutes each. Finally, coverslips were stained with DAPI (Beyotime, C1005, China) in the dark for 10 minutes, mounted with a mounting medium, and observed and photographed under an immunofluorescence microscope.
3.15 Hydrogel's Protective Effect Against ROS in C2C12 Cells
C2C12 cells were seeded in a plate and treated with H2O2 (400 µmol) for 24 hours to establish an oxidative stress microenvironment. Different concentrations of decellularized extracellular matrix materials were then added, and the cells were co-cultured for 48 h. Cell viability was assessed using the CCK-8 and Calcein-AM/PI dual staining method. Intracellular ROS detection was performed using DCFH-DA as the ROS fluorescent probe. Cells were co-incubated with different treatments for 30 minutes, followed by PBS washing. Fluorescence microscopy was used to observe and image the staining of live/dead cells and ROS. Quantitative analysis was conducted using ImageJ software.
3.16 Effect of Hydrogel on Macrophage Polarization
The expression of the macrophage marker CD86 (M1) in RAW cells was detected using flow cytometry. RAW cells were seeded in a 6-well plate at a density of 1×106 cells/mL with 2 mL per well. After 12 h, the medium was replaced with hydrogel extract, and cells were cultured for an additional 3 days. Subsequently, the cells were scraped off, washed twice with phosphate-buffered saline (PBS), and thoroughly resuspended in 250 µL of fixation/permeabilization solution, followed by incubation at 4°C for 20 minutes. After washing the cells twice with buffer (from the fixation/permeabilization kit, BD), they were incubated with antibody solution containing CD86 (dilution factor 1:100, H2316, Santa Cruz Biotech) at 4°C for 30 minutes. After antibody incubation, the cells were washed twice with buffer, resuspended in PBS, and analyzed using a flow cytometer (FACS, AriaII, BD).
3.17 Animal Model Establishment and Grouping
This experiment obtained ethical approval from the Ethics Committee of the Tenth People's Hospital of Shanghai (SHDSYY-2023-3825-3).
The specific steps of the experiment are as follows: After sodium pentobarbital anesthesia of SD rats, an incision was made on the left hind limb posterior lateral thigh to expose the sciatic nerve, freeing the sciatic nerve while preserving the branches of the sciatic nerve to the thigh muscles. The proximal end of the nerve was freed to the level of the hamstring tendon, and the distal end was carefully freed to the entry points of the nerve branches into the muscles. The left sciatic nerve was cut off at the distal end below the hamstring tendon, and the proximal end of the nerve was inverted and buried in the nearby hamstring muscle belly. Two nylon sutures were used to secure the outer membrane and fascicles of the distal end of the nerve, which was pulled proximally and fixed to the hamstring tendon, ensuring that the distal end of the sciatic nerve was positioned at the distal end of the hamstring tendon. Before closing the wound, the left hind limb was maximally moved to ensure secure fixation of the traction lines. The experimental groups were as follows: control group, surgery only without any treatment; material group, injection of UR-gel immediately after surgery without ultrasound treatment; UR-gel + ultrasound group, injection of UR-gel immediately after surgery, followed by 15 minutes of 1 MHz ultrasound stimulation to the tibialis anterior muscle at the 2nd week, with the remaining treatment the same as before; EVs@UR-gel + ultrasound group, injection of EVs@UR-gel immediately after surgery, with the remaining treatment the same as before.
3.18 Measurement of Sciatic Nerve Function Index (SFI)
A wooden trough with open ends, measuring 60 cm in length, 10 cm in width, and 10 cm in height, was constructed. A piece of white paper weighing 70 g was cut to the same length and width as the trough and laid at the bottom. Rats' hind limbs were colored by dipping them in paint at the ankle joints. The rats were then placed at one end of the trough and allowed to walk towards the other end, leaving 5–6 footprints on each side. The following six parameters were measured for each clear footprint: ETS (injured toe spread), NTS (normal toe spread), EPL (injured print length), NPL (normal print length), EIT (injured intermediary toe spread), NIT (normal intermediary toe spread).These indices were then input into the Bain formula to calculate the SFI. An SFI of 0 indicates normal function, while − 100 indicates complete damage. The Bain formula is as follows:
TSF (toe spread factor) = (ETS-NTS)/NTS;
PLF (print length factor) = (EPL-NPL)/NPL;
ITF (intermediary toe spread factor) = (EIT-NIT)/NIT;
SFI = 109.5 TSF − 38.3 PLF + 13.3 ITF − 8.8.
3.19 Measurement of Maximum Isometric Contraction Force
A Kocher needle with a diameter of 1.0 mm was used to secure the distal end of the rat's femur and ankle joint to a wooden board, stabilizing the lower leg segment and maintaining muscle length. Simultaneously, the muscle tendon insertion point of the anterior tibial muscle was surgically exposed. The initial stimulation frequency was set at 10 Hz, with a duration of 0.4 milliseconds, and the voltage was set at 2 V. We calibrated the force transducer using weights of 0 g, 10 g, 20 g, 30 g, and 50 g, respectively. The muscle tendon insertion point of the anterior tibial muscle was connected to the force sensor, and a single stimulation of the proximal end of the sciatic nerve anastomosis was applied using the stimulating electrode, maintaining the aforementioned stimulation parameters. We incrementally increased the weight at the muscle tendon insertion point by 0 g, 5 g, 10 g, 15 g, and 20 g increments (increasing by 5 g each time until the optimal length was determined) to find the maximum force generated by this single stimulation at each weight. The muscle length at this point was considered the optimal length. At the optimal length, the voltage was adjusted to 10 V, and the stimulation frequency was varied at 50 Hz, 100 Hz, 150 Hz, and 200 Hz (starting from 50 Hz and increasing by 50 Hz each time) to measure the maximum isometric contraction force under continuous stimulation of the sciatic nerve at equal lengths.
3.20 Muscle Circumference, Muscle Wet Weight and Histological Examination
The initial length of the muscles at relaxation was measured. Bilateral anterior tibial muscles were excised completely from their origin to insertion points in relaxed muscles. The superficially adherent subcutaneous tissues were carefully removed, then measure the muscle circumference at the thickest part of the muscle and compare it to the unoperated side, and subsequently, the wet weight was measured by using an analytical balance (with a sensitivity of 1 mg, R200D, Germany) and recorded. Frozen sections from the medial heads of the left anterior tibial and gastrocnemius muscles were subjected to Masson's trichrome staining. Acid fuchsin stained muscle fibers red, while aniline blue stained collagen fibers blue.
3.21 Immunofluorescence Staining of Tissue Sections
Tissue sections from the anterior tibial muscle collected at six weeks post-operation were subjected to immunofluorescence staining. The muscle tissue was fixed in 4% formaldehyde, dehydrated through a sucrose gradient, embedded in OCT compound, and sliced into 12-micrometer sections. These sections were then blocked at room temperature for 1 hour. Subsequently, they were incubated overnight at 4°C with primary antibodies, including mouse anti-MyHC (dilution 1:200, Abcam, Cambridge, UK) and rabbit anti-Col-1 (dilution 1:200, Abcam, Cambridge, UK). After rinsing with PBS, the sections were incubated with the corresponding secondary antibodies at room temperature in the dark for 2 hours. Following another round of PBS washing, the sections were stained with DAPI to visualize the cell nuclei. Finally, fluorescence microscopy was employed for imaging.
3.22 Transcriptome Sequencing and Bioinformatics Analysis
At 6th week post-operation, total RNA was extracted from denervated and denervated + EVs@UR-gel anterior tibial muscles using mRNA isolation kit. Subsequently, the RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Ribosomal RNA was enzymatically digested using the TruSeq Stranded Total RNA and Ribo-Zero Gold kits. The fragmented RNA was used as a template for cDNA synthesis and library construction with fragment buffer solution. RNA libraries were then subjected to RNA identification using the Agilent 2100 Bioanalyzer. Sequencing was performed using an Illumina sequencer (HiSeqTM 2500 or Illumina HiSeq X Ten). DESeq software was employed for normalizing mRNA counts for each sample and calculating fold changes. Differential expression of reads between the two groups was assessed using a negative binomial distribution test. Finally, genes with fold changes of either > 1.5 or <-1.5, with a q-value < 0.05, were selected as differentially expressed genes. For functional analysis based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, enrichment was considered significant for p-values < 0.05.
{kind=link}