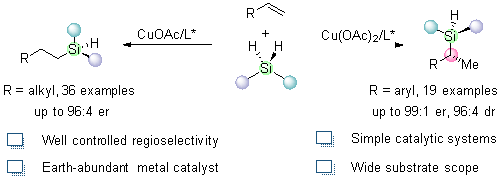
This study presents a copper-catalyzed, substrate-controlled regio- and enantioselective intermolecular hydrosilylation method capable of accommodating a broad scope of alkenes and prochiral silanes. The approach offers an efficient and versatile pathway to generate enantioenriched linear and branched alkyl-substituted Si-stereogenic silanes. Key features of this reaction include mild reaction conditions, simple catalytic systems, compatibility with diverse substrates, high yields, and enantioselectivities.
Article
Copper-catalyzed Intermolecular Regio- and Enantioselective Hy-drosilylation of Alkenes with Prochiral Silanes
https://doi.org/10.21203/rs.3.rs-4743287/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
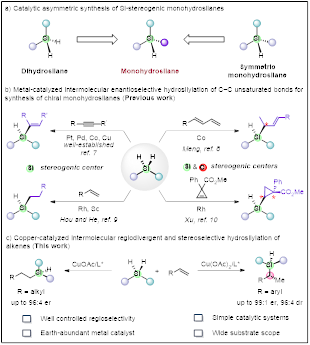
Silicon, like carbon, belongs to Group IVA in the periodic table and is the second most abundant element in the Earth's crust.1 Comparatively, while chiral molecules containing a stereogenic carbon atom have been extensively studied and practically utilized, there has also been substantial interest in synthesizing organic silicon molecules with Si-stereogenic centers.2 These molecules play a pivotal role in various fields such as organic synthesis, functional materials, and biomedicines.3 Traditionally, the creation of silicon-centered chirality has heavily relied on stoichiometric chiral reagents or auxiliaries.4 To enhance efficiency and reduce waste, several novel strategies for synthesizing Si-stereogenic silanes through the catalytic desymmetrization of prochiral organosilanes have been developed (Scheme 1a).5 Additionally, significant advancements have been made recently in constructing silicon-stereogenic silanes from racemic starting materials through dynamic kinetic asymmetric transformation (DYKAT)6a–c and metal-catalyzed kinetic resolution (KR)6d. Among various Si-stereogenic organosilanes, monohydrosilanes have attracted considerable attention due to their unique reactivity and promising applications.3d
Within the realm of strategies developed for the synthesis of chiral monohydrosilanes, metal-catalyzed asymmetric intermolecular hydrosilylation of C–C unsaturated bonds emerges as an efficient approach (Scheme 1b).5, 7–10 It is noteworthy that while enantioselective hydrosilylation of terminal alkenes has been extensively studied for creating a C-stereogenic center with a C-Si bond,3, 5, 11 only limited progress has been made in constructing a Si-stereogenic center via this strategy. To date, there have only been two reported instances of hydrosilylation of terminal olefins for the preparation of linear alkyl-substituted Si-stereogenic silanes.9 In 2018, Hou and colleagues reported a groundbreaking, catalytic, and enantioselective method for synthesizing Si-stereogenic silanes through Sc-catalyzed intermolecular hydrosilylation of terminal alkenes.9a Subsequently, He and co-workers demonstrated an example using Rh-catalysis, yielding Si-stereogenic compounds with moderate enantioselectivities.9b In contrast, only a few intermolecular reaction protocols have successfully enabled the simultaneous creation of both a C- and a Si-stereocenter through noble transition metal-catalyzed enantioselective Si–H bond insertion of carbene species12 or Co-catalyzed intermolecular hydrosilylation of 1,3-dienes8. However, the catalytic asymmetric synthesis of chiral monohydrosilanes with both a C- and a Si-stereogenic center, using readily available alkenes as substrates through the hydrosilylation process, remains unexplored. Therefore, the
development of highly efficient and robust methodologies catalyzed by abundant metals to access chiral hydrosilanes with a Si-stereogenic center or with both a C- and a Si-stereogenic center is highly desirable. Building upon previous work on copper-catalyzed asymmetric hydrosilylation of unsaturated C–C bonds,7e, 11h, 13 we present a Cu-catalyzed intermolecular regio- and enantioselective hydrosilylation of alkenes with prochiral silanes for the synthesis of diverse enantioenriched hydrosilane products (Scheme 1c).
Table 1. Optimization of reaction conditions for the copper-catalyzed hydrosilylation of allylbenzene. a, b, c

a Conditions: 1a (0.2 mmol), 2 (0.6 mmol), copper catalyst (4 mol %) and ligand (8 mol %) were stirred at 40°C for 2 d under N2 atmosphere. b Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. c The er values were determined by chiral HPLC analysis. d 8 mol% catalyst, 8.8 mol% ligand and 8.8 mol% secondary ligand. e 10 mol% catalyst, 11 mol% ligand and 11 mol% secondary ligand. f Isolated yield.
Evaluation of reaction conditions for the copper-catalyzed hydrosilylation of allylbenzene. Using allylbenzene (1a) as the model substrate, the Cu-catalyzed hydrosilylation of alkenes was first investigated by assessing the steric effects of different dihydrosilanes, including PhMeSiH2 (2a), mesityl(methyl)silane (2b), and Ph(tBu)SiH2 (2c). Various copper precursors and ligands were also examined for the model reaction. The results of selected experiments are summarized in Table 1. The reactions between allylbenzene and the dihydrosilanes R1R2SiH2 were conducted using 4 mol% Cu(OAc)2 and 8 mol% (R,R)-Ph-BPE (L1) at 40°C (entries 1 − 3). Notably, it was observed that the reaction of allylbenzene with mesityl(methyl)silane (2b) produced the anti-Markovnikov product at 40°C with moderate conversion and a 94:6 enantiomeric ratio (entry 2). Despite PhMeSiH2 (2a) exhibiting a higher conversion rate compared to mesit-yl(methyl)silane (2b) and Ph(tBu)SiH2 (2c), the latter demonstrated better enantioselectivities. A slight enhancement in enantioselectivity was noted upon transitioning from Cu(OAc)2 to CuOAc as the copper precursor (entry 4). Conversely, the use of CuCl as the catalyst did not yield any reaction (entry 5). By increasing the catalyst loading to 8 mol%, the yield of 3ab could be elevated to 58% (entry 6). Encouragingly, incorporating a secondary ligand proved advantageous for this reaction (entries 7 − 10),14 with CyJohnPhos delivering the optimal outcome (95:5 enanti-omeric ratio; entry 10). Ultimately, the best result was obtained when conducting the reaction of 1a with 2b (3.0 equiv) in the presence of CuOAc (10 mol%), (R,R)-Ph-BPE (L1, 11 mol%), and CyJohnPhos (11 mol%) at 40°C for 2 days, resulting in a 75% yield and a 95:5 enantiomeric ratio (entry 11).
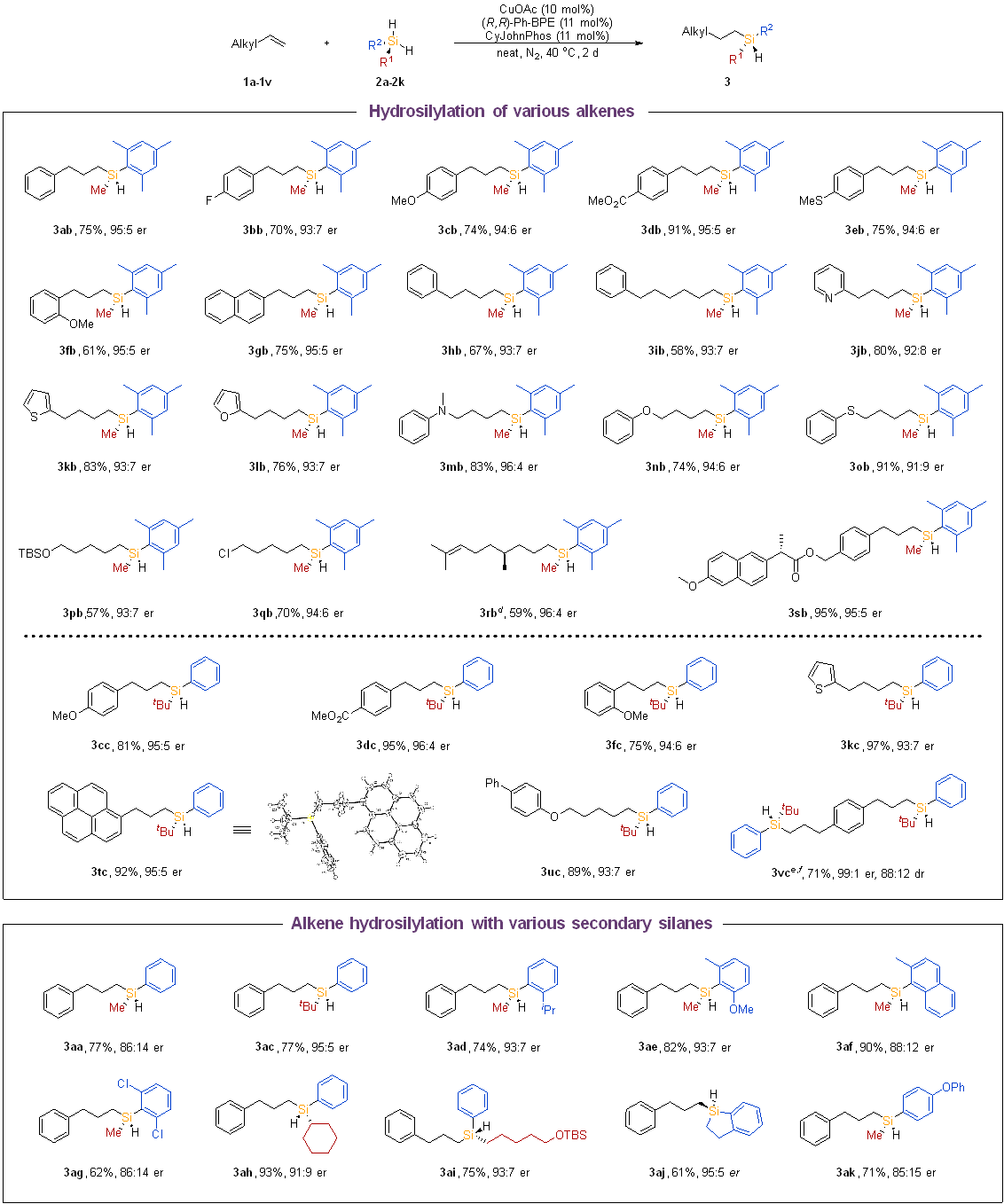
Scope of linear-selective hydrosilylation of alkenes. Following the identification of an active catalyst and optimized conditions for the anti-Markovnikov hydrosilylation of allylbenzene (entry 11 in Table 1), we proceeded to explore the substrate scope. Key findings from this investigation are outlined in Scheme 2. Initially, we examined the hydrosilylation of various alkenes using 2b or 2c. Allylbenzene derivatives containing both electron-donating and electron-withdrawing groups could efficiently undergo reactions with mesityl(methyl)silane (2b) to yield the desired chiral linear products, typically achieving moderate to excellent yields with good enantioselectivities (3ab–3fb).
In general, electron-withdrawing allylbenzene proved to be a superior substrate (3db) that yielded the hydrosilylation product with higher efficiency. The efficiency and enantioselectivity of the desired products were slightly influenced as the carbon chain prolonged, as observed in 3hb and 3ib. Additionally, heteroaryl-substituted alkenes served as suitable substrates, producing chiral silanes with high efficiency and enantioselectivity (3jb–3lb). Various functional groups such as amino (3mb), phenoxy (3nb), thioether (3ob), silyloxy (3pb), halogens (3qb) were all compatible. These reactions proceeded smoothly, yielding the corresponding tertiary silane products with good yields (57–91%) and high enantioselectivities (91:9 to 96:4 er). In cases where the substrate contained both terminal and internal olefin units, the reaction selectively occurred at the less sterically hindered terminal olefin, leaving the internal olefin moiety intact (3rb). When tert-butyl group-substituted silane was utilized, the desired products were obtained with improved efficiency and enantioselectivity (3cc–3uc). The absolute configuration of 3tc was determined through X-ray diffraction analysis (CCDC: 2358701). Furthermore, 1,4-diallylbenzene was selectively hydrosilylated, resulting in the bis-silane product 3vc with a yield of 71%, an enantiomeric ratio of 98:2 er, and a diastereomeric ratio of 88:12 dr. Prochiral silanes were examined under identical conditions (Scheme 2). The efficiency and enantioselectivity of the product were significantly influenced by the steric hindrance of the silane. Reactions involving silanes bearing various bulky aryl or alkyl groups exhibited high enantioselectivity, albeit with a slight decrease in efficiency (3ac–3af). A variety of readily available alkylphenylsilanes proved to be suitable substrates (3ah–3aj). Although the desired products were obtained when the arylmethylsilane contained electron-donating or electron-withdrawing groups (3ag, 3ak), their efficiency and enantioselectivity were negatively affected.
Evaluation of reaction conditions for the copper-catalyzed hydrosilylation of styrene. Based on the aforementioned investigation, we aimed to expand the substrate scope by incorporating aryl alkenes (Table 2). Despite compound (R,R)-5ak demonstrating excellent efficiency and a favorable enantiomeric ratio, the diastereomeric ratio achieved under the optimized conditions (entry 11, Table 1) was only moderately satisfactory (entry 1). It is worth noting that introducing a second ligand in this reaction didn’t have a positive effect (entry 2). When switching from CuOAc to Cu(OAc)2 as the copper precursor (entry 3), a slight increase in diastereoselectivity was observed. Although different chiral ligands were tested, the desired outcomes were not achieved (entries 4–6). En-couragingly, increasing the amounts of the chiral ligand yielded positive results (entries 7–8). Reducing the reaction time to 36 hours did not significantly alter the results, resulting in the target product (R,R)-5ak being obtained in a 91% isolated yield, with a 98:2 er and up to a 95:5 dr ratio (entry 9).
Table 2. Optimization of reaction conditions for the copper-catalyzed hydrosilylation of allylbenzene. a, b, c, d

a Conditions: 4a (0.2 mmol), 2k (0.6 mmol), copper catalyst (4 mol%) and ligand were stirred at 40°C for 2 d under N2 atmosphere. b Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard. c The dr values were determined by GC analysis of the crude reaction mixture. d The er values were determined by chiral HPLC analysis. e 10 mol% CuOAc. f 36 h. g 24 h. h Isolated yield.
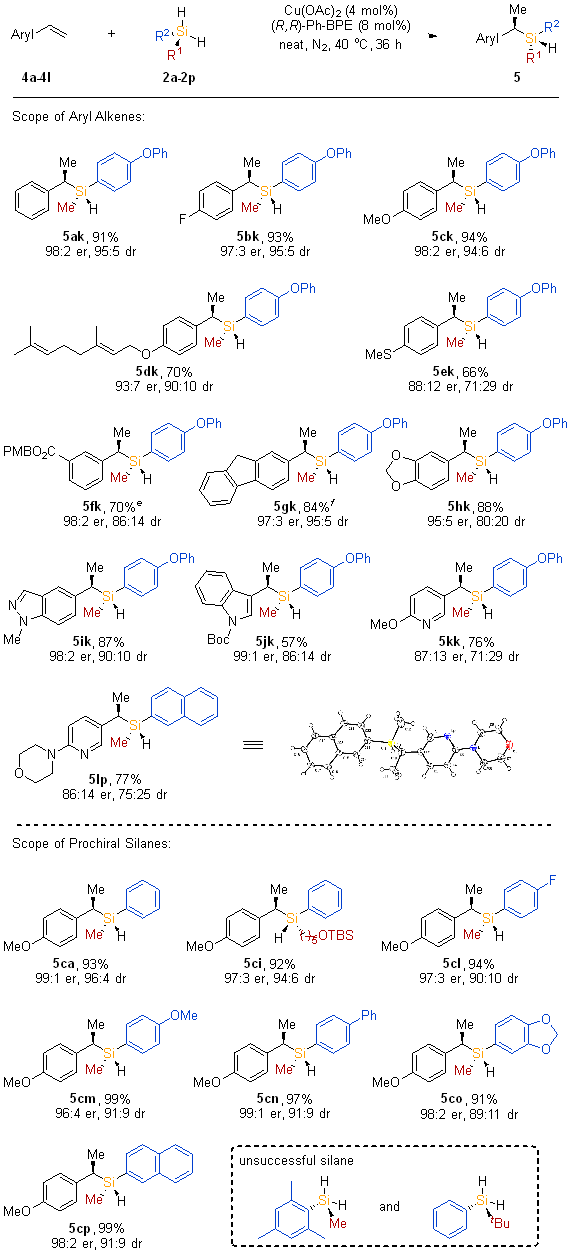
Scope of branched-selective hydrosilylation of alkenes. Under the optimized conditions, we explored the substrate scope as illustrated in Scheme 3. Enantioenriched branched silanes were successfully synthesized, incorporating halo-genated (5bk), electron-rich (5ck), electron-deficient (5fk) aryl groups, or a fused aromatic ring (5gk). Yields varied between 70% and 94%, with diastereomeric ratios ranging from 86:14 to 95:5, and enantiomeric ratios reaching up to 98:2. Notably, the reaction did not accommodate aryl bromide and iodide substrates. A styrene derivative carrying a methylthio group (5ek) was obtained in moderate yield but displayed poor diastereo- and enantioselectivity. Of significance was the selective transformation of aryl alkenes bearing trisubstituted olefin moieties with the silane reagent resulting in high efficiency and stereoselectivity (5dk). Furthermore, a wide array of heteroaryl-substituted alkenes proved to be suitable substrates for the production of chiral silanes exhibiting both C- and Si-stereogenic cen-ters efficiently, enantioselectively, and with moderate diastereoselectivity (5ik–5lp). The absolute configuration of the chiral branched alkylsilane product was conclusively determined through X-ray crystallographic analysis of compound 5lp (CCDC: 2358711).
Next, the investigation focused on the scope of prochiral silanes. Reactions with readily available arylmethylsilanes exhibited remarkable efficiency, diastereoselectivity, and enantioselectivity (5ca, 5cl–5cp). Another alkylarylsilane (5ci) was converted to the target products with a yield of 92%, a diastereomeric ratio (dr) of 94:6, and an enantiomeric ratio (er) of 97:3. However, diarylsilanes and sterically hindered silanes such as mesityl(methyl)silane (2b) and Ph(tBu)SiH2 (2c) were found to be unsuitable substrates for these reactions.
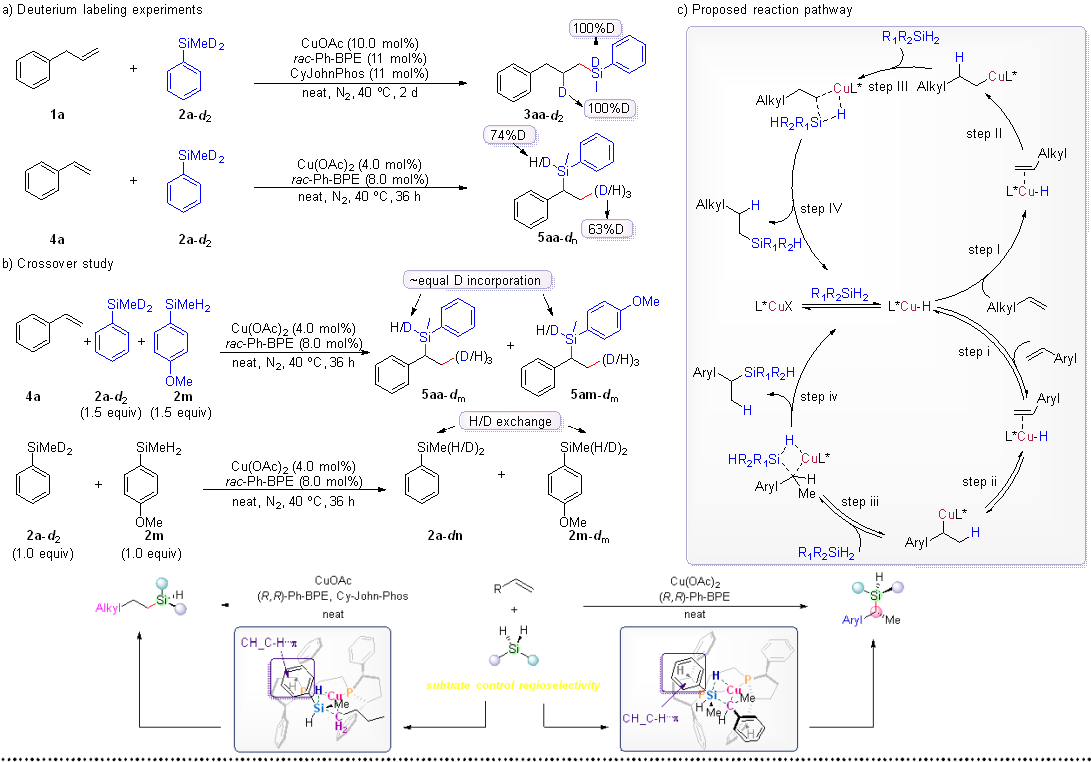
Mechanistic Investigation. Deuterium labeling experiments were conducted as part of the investigation into the reaction mechanism. Initially, the isotopically labeled substrate methyl(phenyl)silane-d2 (2a-d2) was subjected to standard conditions, leading to the formation of deuterated products 3aa-d2 and 5aa-dn, illustrated in Scheme 5a. To enhance our comprehension of the reversibility factors that influence the reaction steps, multiple control experiments were performed. When styrene (4a), 2a-d2, and (4-methoxyphenyl)(methyl)silane (2m) were simultaneously employed under standard conditions, the integration of the Si- H/D peaks of 5aa-dm and 5am-dm gives a ratio of approximately 1:1. The obtained result provides further evidence for the reversibility of migratory insertion and β-hydride elimination.15 Another crossover experiment was performed using 1.0 equiv 2a-d2 and 2m reacting without an alkene under standard conditions. The presence of deuterium crossover was confirmed through 1H NMR analysis. These findings strengthen our conclusion that the generation of copper hydride species, migratory insertion, and β-hydride elimination display reversibility (Scheme 5b). Based on this data, we propose a potential reaction mechanism for the copper-catalyzed intermolecular regiodivergent and stereoselective hydrosilylation of alkenes (Scheme 5c).
Computational studies. To elucidate the origin of the regio- and stereoselectivities observed in the reaction, we resorted to DFT studies on the hydride-insertion and the subsequent metathesis steps (See SI for the details). The potential energy surface leading to the linear products was explored with 1-butene as the model substrate (Scheme 6a). The migratory insertion step from S1 to TS1 has a barrier of 10.9 kcal/mol, generating the linear alkyl Cu(I) P1, with an energy downhill of 23.1 kcal/mol. Subsequent silane association leads to further energy downhill of 3.3 kcal/mol. From S2, the conformation space of the metathesis step was mapped similarly. Our calculation shows that the most energetically favored pathway proceeds through transition structure TS2_conf B_R (in favor of the R-product) with a barrier of only 13.4 kcal/mol, which is much more favored (by 13.0 kcal/mol) than the β-H elimination backward pathway (26.4 kcal/mol), in congruent with the absence of H/D scrambling observed experimentally (Scheme 5a). The second lowest-energy TS is TS2_conf A_S which is 3.8 kcal/mol higher, in good agreement of the sense and degree of enantiocontrol observed experimentally.
For the reaction with styrene as starting material, we systematically sampled the conformational space of the hydrocupration and metathesis transition states in the formation of the branched product. It was found that the relative energy of the conformational of the R configuration was lower than that of the S configuration, where TS1’_conf 2_re was the conformation with the lowest energy (Scheme 5b). The results show that the Cu–H insertion TS forming terminal C–Cu bond (TS1’_r.r.) is 8.8 kcal/mol higher than TS1’_conf 2_re, which is consistent with the exclusive regioselectivity observed experimentally. The most stable conformation TS1’_conf 2_re leads to the lowest energy benzyl-Cu(I) intermediate P1’_conf 2_re, which then associates with the phenyl silane to give the σ-complex S2’ with a 3.8 kcal/mol energy drop. Subsequently, the conformational space of the metathesis with Ph(Me)SiH2 was also mapped. Of these transition structures, TS2’_conf b_R,R and TS2’_conf d_R,R converged to the same structure which was found to be lowest in energy. Notably, there is only a small energy difference of 1.9 kcal/mol between the β-H elimination from the benzylic Cu(I) and the metathesis, suggesting of a partially reversible hydrometallation step before the rate-determining metathesis. This can explain the isotope scrambling observed in the mechanistic experiments (Scheme 5a, 5b). According to distortion interaction analysis (DIAS) of these TSs, it can be concluded that for both substrates, the configuration established at the silicon atom are a result of differences of the distortion of the silane moiety.
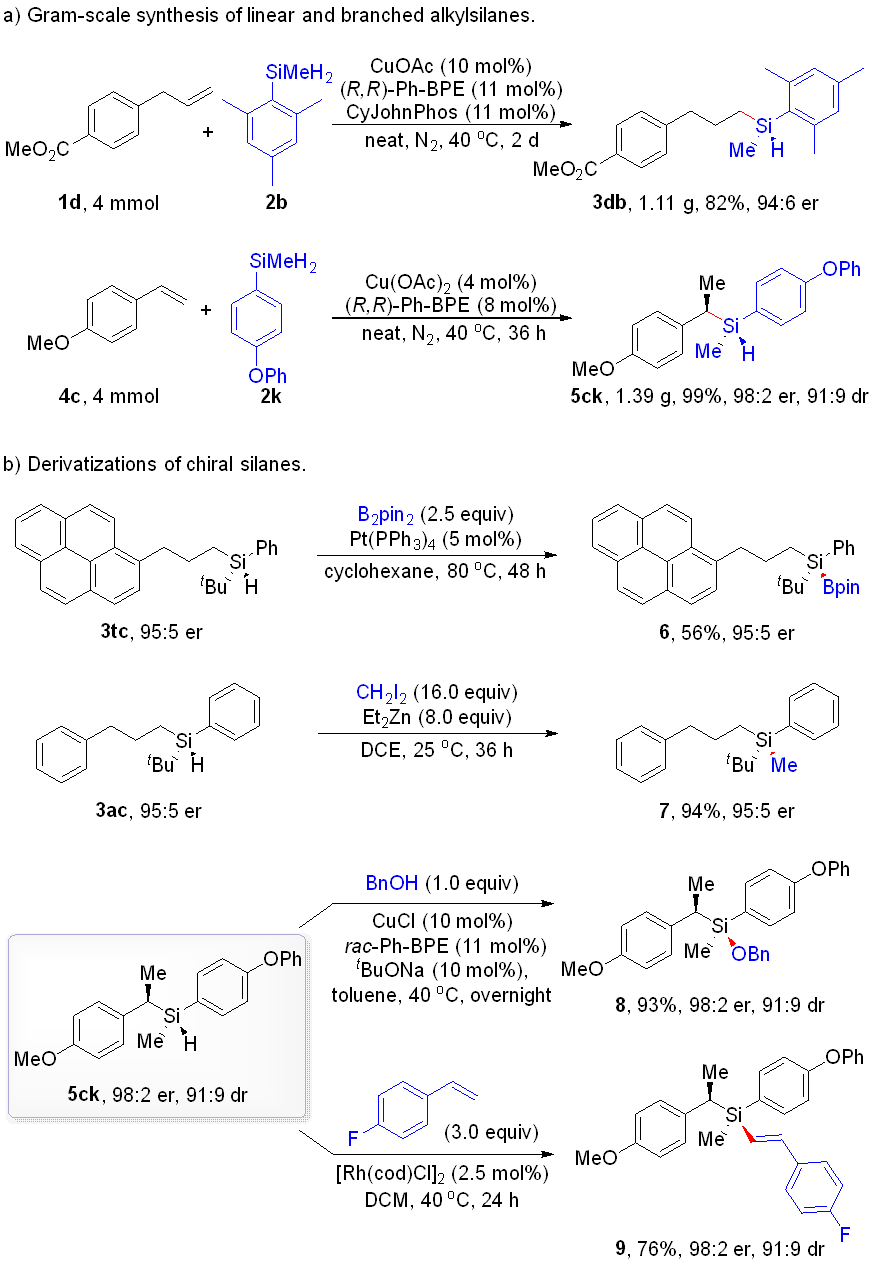
In summary, we have successfully developed a copper-catalyzed, highly selective hydrosilylation method for alkenes. Various monosubstituted alkenes with aromatic and aliphatic groups efficiently reacted with hydrosilanes to produce enantiomerically enriched alkyl-substituted silanes with a Si-stereogenic center or Si/C two stereogenic centers under substrate influence. Control experiments suggested that metathesis likely plays a crucial role as the rate-determining step. The outstanding regioselectivity and enantioselectivity were further confirmed through DFT calculations. Additionally, the unreacted Si − H bond in the chiral silane products provides opportunities for further derivatization.
Author contributions
Y.-H. Xu directed the project and composed the manuscript with revisions provided by the other authors. X.-Y. Zhu, J.-L. Xu and Z.-L. Wang performed the experiments. W. Gao and J.-B. Zhao performed the DFT calculations. All the authors were involved the analysis of results and discussions of the project.
Competing interests
The authors declare no competing interests.
Acknowledgment
We gratefully acknowledge research support of this work by the funding of the National Natural Science Foundation of China (22371269), the State Key Laboratory of Elemento-organic Chemistry Nankai University (202001), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB0450301), the Open Project of Key Laboratory of Organosilicon Chemistry, and Material Technology of Ministry of Education, Hangzhou Normal University (KFJJ2022013).
- Tréguer P, Nelson DM, Bennekom AJV, DeMaster DJ, Leynaert A, Quéguiner B The Silica Balance in the World Ocean, Struyf E, Smis A, Van Damme S, Meire P, Conley DJ The Global Biogeochemical Silicon Cycle (eds) (1995) : A Rees-timate. Science 268, 375 (b) Struyf E, Smis A, Van Damme S, Meire P, Conley DJ The Global Biogeochemical Silicon Cycle. Silicon 1, 207 (2009)
- Schwarz J (2016) Atypical Elements in Drug Design. Springer, Heidelberg
- (a) Igawa K, Tomooka K Chiral Silicon Molecules, In Organosilicon Chemistry: Novel Approaches and Reactions, Hiyama E, T., Oestreich M, Wiley, Weinheim, Germany, pp. 495–532 (b), Xu L-W, Li L, Lai G-Q, Jiang J-X (2019) The Recent Synthesis and Application of Silicon-Stereogenic Silanes: A Renewed and Significant Challenge in Asymmetric Synthesis. Chem. Soc. Rev. 40, 1777–1790 (2011). (c) Bauer, J. O. & Strohmann, C. Recent Progress in Asymmetric Synthesis and Application of Difunctionalized Silicon-Stereogenic Silanes. Eur. J. Inorg. Chem. 2016, 2868–2881 (2016). (d) Wu, Y. & Wang, P. Silicon-Stereogenic Monohydrosilane: Synthesis and Applications. Angew. Chem. Int. Ed. 61, e202205382 (2022)
- (a) Sommer LH, Stereochemistry, Mechanism and (b) Kipping, F. S. XXII. Organic Derivatives of Silicon. Part II. The Synthesis of Benzylethylpropylsilicol, its Sulphonation, and the Resolution of the Dl-sulphonic Derivative into its Optically Active Components J. Chem. Soc. 91, 209–240 (1907). (c) Sommer, L. H. & Silicon; an Introduction to the Dynamic Stereochemistry and Reaction Mechanisms of Silicon Centers; McGraw-Hill: New York, Frye CL, Rendler S, Auer G, Oestreich M Kinetic Resolution of Chiral Secondary Alcohols by Dehydrogena-tive Coupling with Recyclable Silicon-stereogenic Silanes., Rendler S, Oestreich M, Butts CP, Lloyd-Jones GC (1965) Optically Active Organosilicon Compounds Having Reactive Groups Bonded to Asymmetric Silicon. Displacement Reactions at Silicon with Pure Retention and Pure Inversion of Configuration. J. Am. Chem. Soc. 81, 4, 1013 (1959). (d) Yamamoto, K., Kawanami, Y. & Miyazawa, M. Synthesis of a Highly Enantiomerically Enriched Silyllithium Compound. J. Chem. Soc. Chem. Commun. 436 (1993). (e) Strohmann, C., Hörnig, J., & Auer, D. Synthesis of a Highly Enantiomerically Enriched Silyllithium Compound. Chem. Commun. 2, 766–767 (2002). (f) Trzoss, M., Shao, J. & Bienz, S. Preparation of a ‘Si-centered’ Chiral Auxiliary by Resolution. Tetrahedron Asymmetry 15, 1501–1505 (2004). (g) Rendler, S., Auer, G., Oestreich, M. Kinetic Resolution of Chiral Secondary Alcohols by Dehydrogena-tive Coupling with Recyclable Silicon-stereogenic Silanes. Angew. Chem. Int. Ed. 44, 7620–7624 (2005). (h) Rendler, S., Auer, G., Keller, M. & Oestreich, M. Preparation of a Privileged Silicon-stereogenic Silane: Classical Versus Kinetic Resolution. Adv. Synth. Catal. 348, 1171–1182 (2006). (i) Rendler, S., Oestreich, M., Butts, C. P., Lloyd-Jones, G. C. Intermolecular Chirality Transfer from Silicon to Carbon: Interrogation of the Two-silicon Cycle for Pd-catalyzed Hydrosilylation by Stereoisotopochemical Crossover. J. Am. Chem. Soc. 129, 502–503 (2007). (j) Igawa, K., Takada, J., Shimono, T. & Tomooka, K. Enantioselective Synthesis of Silanol. J. Am. Chem. Soc. 130, 16132–16133 (2008). (k) Igawa, K., Kokan, N. & Tomooka, K. Symmetric Synthesis of Chiral Silacarboxylic Acids and Their Ester Derivatives. Angew. Chem. Int. Ed. 49, 728–731 (2010)
- (b) For reviews, see: (a) Xu, L.-W. Desymmetrization Catalyzed by Tran-sition-Metal Complexes: Enantioselective Formation of Silicon-Stereogenic Silanes. Angew. Chem. Int. Ed. 51, 12932–12934, Shintani R, Recent Advances in the Transition-Metal‐Catalyzed Enantioselective Synthesis of Silicon-Stereogenic Organosilanes, Xu L-W, Huang X, Ke J, He C, Zeng Y, Ye F (2012). Asian J. Org. Chem. 4, 510–514 (2015). (c) Cui, Y.-M., Lin, Y. & Xu, L.-W. Catalytic Synthesis of Chiral Organoheteroatom Compounds of Silicon, Phosphorus, and Sulfur via Asymmetric Transition Metal-Catalyzed C–H Functionalization. Coord. Chem. Rev. 330, 37–52 (2017). (d) Shintani, R. Recent Progress in Catalytic Enantioselective Desymmetrization of Prochiral Organosilanes for the Synthesis of Silicon-Stereogenic Compounds. Synlett 29, 388–396 (2018). (e) Ye, F. & Xu, L.-W. A Glimpse and Perspective of Current Organosilicon Chem-istry from the View of Hydrosilylation and Synthesis of Silicon-Stereogenic Silanes. Synlett 32, 1281–1288 (2021). (f) Zheng, L., Nie, X.-X., Wu, Y. & Wang, P. Construction of Si-Stereogenic Silanes through C – H Activation Approach. Eur. J. Org. Chem. 44, 6006–6014 (2021). (g) He, C. & Yuan, W. Enantioselective C–H Functionalization toward Sili-con-Stereogenic Silanes. Synthesis 54, 1939–1950 (2022). (h) Ge, Y., Huang, X., Ke, J., He, C. Transition-metal-catalyzed Enantioselective C-H silylation. Chem Catal. 2, 2898–2928 (2022). (i) Xu, L.-W., Huang, W.-S., Wang, Q. & Yang, H. State-of-the-art Advances in Enantioselective Transition-Metal-Mediated Reactions of Silacyclobutanes. Synthesis 54, 5400–5408 (2022). (j) Zeng, Y., Ye, F. Research Progress on New Catalytic Reaction Systems for Asymmetric Synthesis of Silicon-Stereogenic Center Containing Compounds. Chin. J. Org. Chem. 43, 3388–3413 (2023). (k) Li, L., Huang, W., Xu, Z. & Xu, L. Catalytic Asymmetric Silicon-carbon Bond-forming Transformations Based on Si-H Functionalization. Sci. China Chem. 66, 1654–1687 (2023). (l) Zhao, J., Ge, Y. & He, C. Construction of Silicon-Stereogenic Center via Catalytic Asymmetric Si-H/X-H Dehydrogenative Coupling. Chin. J. Org. Chem. 43, 3352–3366 (2023)
- (a) Zeng Y, Fang X-J, Tang R-H, Xie J-Y, Zhang F-J, Xu Z, Nie Y-X, Xu L-W Rhodium-Catalyzed Dynamic Kinetic Asymmetric Hydrosi-lylation toAccess Silicon-Stereogenic Center. Angew. Chem. Int. (ed). 61, e202214147 (b) Zhou H, Properzi R, Leutzsch M, Belanzoni P, Bistoni G, Tsuji N, Han JT, Zhu C, List B, Gou F-H, Ren F, Wu Y-C, Wang P Catalytic Kinetic Resolution of Monohydrosilanes via Rhodium-Catalyzed Enantioselective Intramolecular Hydrosilylation. (2022) Organocatalytic DYKAT of Si-Stereogenic Silanes. J. Am. Chem. Soc. 145, 4994–5000 (2023). (c) Hu, T., Zhao, C., Zhang, Y., Kuang, Y., Gao, L., Wang, W., Su, Z. & Song, Z. Enantioconvergent Construction of Stereogenic Silicon via Lewis Base-Catalyzed Dynamic Kinetic Silyletherification of Racemic Chlorosilanes. Nat. Commun. 14, 4900 (2023). (d) Gou, F.-H., Ren, F., Wu, Y.-C., Wang, P. Catalytic Kinetic Resolution of Monohydrosilanes via Rhodium-Catalyzed Enantioselective Intramolecular Hydrosilylation. Angew. Chem. Int. Ed 63, e202404732 (2024)
- (b) Selected examples for enantioselective hydrosilylation of alkynes, see: (a) Igawa, K., Yoshihiro, D., Ichikawa, N., Kokan, N. & Tomooka, K. Catalytic Enantioselective Synthesis of Alkenylhydrosilanes. Angew. Chem. Int. Ed. 51, 12745–12748, Wen H, Wan X, Huang Z Asymmetric Synthesis of Silicon-Stereogenic Vinylhydrosilanes by Cobalt-Catalyzed Regio- and Enantioselective Alkyne Hydrosilylation with Dihydrosilanes., Ling F, Ye F, Fang X, Zhou X, Huang W, Xu Z, Xu L (2012) 57, 6319–6323 (2018). (c) Xie, J.-L., Xu, Z., Zhou, H.-Q., Nie, Y.-X., Cao, J., Yin, G.-W., Bouillon, J.-P. & Xu, L.-W. Palladium-catalyzed Hydrosilylation of Ynones to Access Silicon-stereogenic Silylenones by Stereospecific Sromatic Interaction-assisted Si-H Activation. Sci. China Chem. 64, 761–769 (2021). (d) Ling, F., Ye, F., Fang, X., Zhou, X., Huang, W., Xu, Z., Xu, L. An Unusual Autocatalysis with an Air-stable Pd Complex to Pro-mote Enantioselective Synthesis of Si-stereogenic Enynes. Chem. Sci. 14, 1123–1131 (2023). (e) Xu, J., Wang, Z., Zhao, J. & Xu, Y. Enantioselective Construction of Si-stereogenic Linear Alkenylhydrosilanes via Copper-catalyzed Hydrosilylation of Alkynes. Chem Catal. 4, 100887 (2024)
- Wang L, Lu W, Zhang J, Chong Q, Meng F (2022) Cobalt-Catalyzed Regio-, Diastereo- and Enantioselective Intermolecular Hydrosilylation of 1,3-Dienes with Prochiral Silanes. Angew Chem Int Ed 61:e202205624
- Zhan G, Teng HL, Luo Y, Lou SJ, Nishiura M, Hou Z Enantioselective Construction of Silicon-Stereogenic Silanes by Scandium-Catalyzed Intermolecular Alkene Hydrosilylation., He T, Liu L-C, Ma W-P, Li B, Zhang Q-W, He W Enantioselective Construction of Si-Stereogenic Center (eds) (2018) 57, 12342–12346 (b) He T, Liu L-C, Ma W-P, Li B, Zhang Q-W, He W Enantioselective Construction of Si-Stereogenic Center via Rhodium-Catalyzed Intermolecular Hydrosilylation of Alkene. Chem. Eur. J. 26, 17011–17015 (2020)
- Zhao Z-Y, Nie Y-X, Tang R-H, Yin G-W, Cao J, Xu Z, Cui Y-M, Zheng Z-J, Xu L-W (2019) Enantioselective Rhodium-Catalyzed Desymmetric Hydrosilylation of Cyclopropenes. ACS Catal 9:9110–9116
- (b) Uozumi, Y. & Hayashi, T. Catalytic Asymmetric Synthesis of Optically Active 2-alkanols via Hydrosilylation of 1-alkenes with a Chiral Monophosphine-palladium Catalyst. J. Am. Chem. Soc. 113, 9887–9888 (1991). (c) Selected examples for enantioselective hydrosilylation of terminal alkenes, see: (a) Gibson, S. E. & Rudd, M. The Role of Secondary Interactions in the Asymmetric Palladium-Catalysed Hydrosilylation of Olefins with Monophosphane Ligands. Adv. Synth. Catal. 349, 781–795, Jensen JF, Svendsen BY, Cour I, Pedersen TV, H. L., Jo-hannsen M, Junge K, Wendt B, Enthaler S, Beller M, Cheng B, Liu W, Lu Z, Chen C, Wang H, Sun Y, Cui J, Xie J, Shi Y, Yu S, Hong X, Lu (2007) Highly Enantioselective Hydrosilylation of Aromatic Alkenes. J. Am. Chem. Soc. 124, 4558–4559 (2002). (d) Guo, X.-X., Xie, J.-H., Hou, G.-H., Shi, W.-J., Wang, L.-X. & Zhou, Q.-L. Asymmetric Palladium-catalyzed Hydrosilylation of Styrenes Using Efficient Chiral Spiro Phosphoramidite Ligands. Tetrahedron: Asymmetry 15, 2231–2234 (2004). (e) Junge, K., Wendt, B., Enthaler, S., Beller, M. Palladium-Catalyzed Enantioselective Hydrosilylation of Aromatic Olefins. ChemCatChem 2, 453–458 (2010). (f) Weber, I. & Jones, G. B. Bidentate Planar Chiral η6-arene Tricarbonyl Chromium(0) Complexes: Ligands for Catalytic Asymmetric Alkene Hydrosilylation. Tetrahedron Lett. 42, 6983–6986 (2001). (g) Zhang, F. & Fan, Q.-H. Synthesis and Application of Bulky Phosphoramidites: Highly Effective Monophosphorus Ligands for Asymmetric Hydrosilylation of Styrenes. Org. Biomol. Chem. 7, 4470–4474 (2009). (h) Gribble, M. W., Jr., Pirnot, M. T., Bandar, J. S., Liu, R. Y. & Buchwald, S. L. Asymmetric Copper Hydride-Catalyzed Markovnikov Hydrosilylation of Vinylarenes and Vinyl Heterocycles. J. Am. Chem. Soc. 139, 2192 – 2195 (2017). (i) Naito, T., Yoneda, T., Ito, J. & Nishiyama, H. Enantioselective Hydrosilylation of Aromatic Alkenes Catalyzed by Chiral Bis(oxazolinyl)phenyl–Rhodium Acetate Complexes. Synlett 23, 2957–2960 (2012). (j) Kitayama, K., Uozumi, Y. & Hayashi, T. Palladium-catalysed Asymmetric Hydrosilylation of Styrenes with a New Chiral Monodentate Phosphine Ligand. J. Chem. Soc. Chem. Commun. 1533–1534 (1995). (k) Cheng, B., Liu, W., Lu, Z. Iron-Catalyzed Highly Enantioselective Hydrosilylation of Unactivated Terminal Alkenes. J. Am. Chem. Soc. 140, 5014–5017 (2018). (l) Cheng, B., Lu, P., Zhang, H., Cheng, X. & Lu, Z. Highly Enantioselective Cobalt-Catalyzed Hydrosilylation of Alkenes. J. Am. Chem. Soc. 139, 9439–9442 (2017). (m) Chen, J., Cheng, B., Cao, M. & Lu, Z. Iron-Catalyzed Asymmetric Hydrosilylation of 1,1-Disubstituted Alkene. Angew. Chem. Int. Ed. 54, 4661–4664 (2015). (n) Wen, H., Wang, K., Zhang, Y., Liu, G. & Huang, Z. Cobalt-Catalyzed Regio- and Enantioselective Markovnikov 1,2-Hydrosilylation of Conjugated Dienes. ACS Catal. 9, 1612–1618 (2019). (o) Chen, C., Wang, H., Sun, Y., Cui, J., Xie, J., Shi, Y., Yu, S., Hong, X., Lu Z. Iron-Catalyzed Asymmetric Hydrosilylation of Vinyl-cyclopropanes via Stereospecific C-C Bond Cleavage. iScience 23, 100985 (2020). (p) You, Y. & Ge, S. Asymmetric Cobalt-Catalyzed Regiose-lective Hydrosilylation/Cyclization of 1,6-Enynes. Angew. Chem. Int. Ed. 60, 12046–12052 (2021)
- Yasutomi Y, Suematsu H, Katsuki T Iridium(III)-Catalyzed Enantioselective Si – H Bond Insertion and Formation of an Enanti-oenriched Silicon Center, Jagannathan JR, Fettinger JC, Shaw JT, Franz AK Enantioselective Si–H Insertion Reactions of Diarylcarbenes for the Synthesis of Silicon-Stereogenic Silanes (eds) (2010) J. Am. Chem. Soc. 132, 4510–4511 (b) Jagannathan JR, Fettinger JC, Shaw JT, Franz AK Enantioselective Si–H Insertion Reactions of Diarylcarbenes for the Synthesis of Silicon-Stereogenic Silanes. J. Am. Chem. Soc. 142, 11674–11679 (2020)
- (a) Nishino S, Hirano K, Miura M Catalyzed Reductive gem-Difunctionalization of Terminal Alkynes via Hydrosilylation/ Hydroamination Cascade: Concise Synthesis of α-Aminosilanes. Chem. Eur J. 26, 8725–8728 (b) Wang H, Zhang G, Zhang Q, Wang Y, Li Y, Xiong T, Zhang M, Ji Y, Zhang Z, Zhang C (2020) Copper-Catalyzed Non-directed Hydrosilylation of Cyclopropenes: Highly Diastereoselective Synthesis of Fully Substituted Cyclopropylsilanes. Chem. Commun. 56, 1819 – 1822 (2020). (c) Xu, Q.-F., Yang, P., Zhang, X. & You, S.-L. Enantioselective Synthesis of 4-Silyl-1,2,3,4-tetrahydroquinolines via Copper(I) Hydride Catalyzed Asymmetric Hydrosilylation of 1,2-Dihydroquinolines. Synlett 32, 505–510 (2021). (d) Xu, J.-L., Xu, Z.-Y., Wang, Z.-L., Ma, W.-W., Sun, X.-Y., Fu, Y. & Xu, Y.-H. Copper-catalyzed Regiodivergent and Enantioselective Hydrosilylation of Allenes. J. Am. Chem. Soc. 144, 5535–5542 (2022). (e) Zhang, M., Ji, Y., Zhang, Z., Zhang, C. Copper-Catalyzed Highly Selective Hydrosilylation of Silyl or Boryl Alkene: A Method for Preparing Chiral Geminated Disilyl and Borylsilyl Reagents. Org. Lett. 24, 2756–2761 (2022). (f) Li, S., Xu, J.-L. & Xu, Y.-H. Copper-Catalyzed Enantioselective Hydrosilylation of Allenes to Access Axially Chiral (Cyclohexylidene)ethyl Silanes. Org. Lett. 24, 6054–6059 (2022). (g) Wang, Z., Li, Q., Yang, M., Song, Z., Xiao, Z., Ma, W., Zhao, J. & Xu, Y. Regio- and Enantioselective CuH-catalyzed 1,2- and 1,4-Hydrosilylation of 1,3-Enynes. Nat. Commun. 14, 5048 (2023). (h) Jin, C., He, X., Chen, S., Guo, Z., Lan, Y. & Shen, X. Axial chirality reversal and enantioselective access to Si-stereogenic silylallene. Chem 9, 2956–2970 (2023)
- Lipshutz BH, Noson K, Chrisman W, Lower A (2003) Asymmetric Hydrosilylation of Aryl Ketones Catalyzed by Copper Hydride Complexed by Nonracemic Biphenyl Bis-phosphine Ligands. J Am Chem Soc 125:8779
- Rix FC, Brookhart M, White PS (1996) Electronic Effects on the β-Alkyl Migratory Insertion Reaction of Para-Substituted Styrene Methyl Palladium Complexes. J Am Chem Soc 118:2436–2448
Schemes 1 to 5 are available in the Supplementary Files section
There is NO Competing Interest.
- Scheme1.png
Scheme 1. Synthesis of Silicon-stereogenic monohydrosilanes.
- Scheme2.png
Scheme 2. Scope of the borylation reaction of aryl and aliphatic substituted allylsilanes.a, b, c a Conditions: 1 (0.2 mmol, 1.0 equiv), 2 (3.0 equiv), CuOAc (10 mol %), (R,R)-Ph-BPE (11 mol %) and CyJohnPhos (11 mol %) were stirred at 40 °C for 2 d under N2 atmosphere. (See Supporting Information for the detailed experimental procedures). b Isolated yields. c The er val-ues were determined by chiral HPLC analysis. d 4 d. e 2c (6.0 equiv) was added. f The dr value was determined by chiral HPLC analysis.
- Scheme3.png
Scheme 3. Scope of branched-selective hydrosilylation of alkenes. a, b, c, d a Conditions: 4 (0.2 mmol, 1.0 equiv), 2 (3.0 equiv), Cu(OAc)2 (4.0 mol%) and (R,R)-Ph-BPE (8.0 mol%) were stirred at 40 °C for 36 h under N2 atmosphere. (See Supporting Information for the detailed experimental procedures). b Isolated yields. c The er values were determined by chiral HPLC analysis. d The dr values were determined by GC analysis or 1H NMR of the crude reaction mixture. e 72 h. f In extra dry cyclohexane (2.0 M).
- Scheme4.png
Scheme 4. Gram-scale synthesis and functionalization.
- Scheme5.png
Scheme 5. Mechanistic studies and proposed reaction pathway.
- AdditionalFigure.png
- supportinginformation.pdf
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}