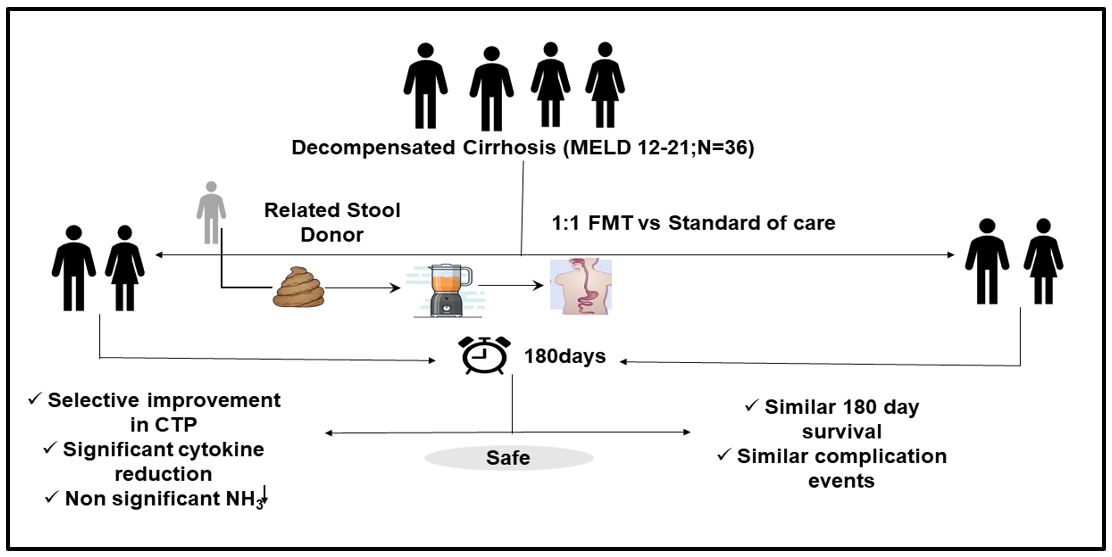
Conduct of the study and study design
The study was an investigator-initiated, open-label randomized clinical trial at a tertiary care referral hospital with a specialized liver unit carried out between August 2018 to November 2019. Approval was obtained before the commencement of the study from the institutional ethics committee (INT/IEC2018/2076) and the trial was registered (ClinicalTrials.gov number, NCT04842539). The trial adhered to the CONSORT guidelines for randomized controlled trials, provisions of the Declaration of Helsinki, and good clinical practice guidelines. Informed consent was obtained prior to enrollment from each patient/relative and a stool donor after an appropriate screening. All the study authors had access to the trial data and approved the final manuscript.
Patient Selection
Consecutive inpatients and outpatients with DC were screened for enrollment eligibility. Patients satisfying the selection criteria were enrolled in the study.
Inclusion Criteria
Patients in the age group of 18-65 years with a diagnosis of DC (of any etiology) based on clinical, radiological, or histological criteria with model for end-stage liver disease (MELD scores) between 12-21 were included.
Exclusion criteria
Patients with an ongoing bacterial infection requiring antibiotics or those having received antibiotics/pre-pro biotics within the last 14 days, those with a history of significant alcohol intake in the previous two months, those with a recent (<14 days) history of spontaneous bacterial peritonitis, HE or variceal bleed, patients with a history of substance abuse or psychiatric illness, those with HIV infection, pregnant patients, patients with hepatocellular carcinoma or other known malignancy, those with history of prior liver transplantation or bariatric surgery, or those on immunosuppression, those with a history suggestive of inflammatory bowel disease, celiac disease, history of allergy to food substances were excluded.
Study objectives
Primary Objective: To assess the difference in 180-day mortality between the FMT group and the SOC group.
Secondary Objective
To assess and compare the changes in CTP, MELD, and MELD Na scores (day 28, 90, and 180 ) and to assess the changes in ammonia levels (day 7 and day 28) and inflammatory markers (IL-1b, IL-6, ) at day 28 between FMT and SOC group.
Randomization of patients
After satisfying the selection criteria were randomized into two groups 1:1 ratio by an unrelated person using a computer-generated random number table. Allocation concealment was done using sequentially numbered opaque sealed envelopes. The physician administering FMT was aware of the treatment being administered, as the nature of the intervention meant that it was not possible to make an identical placebo.
Group 1 (FMT Group): Patients with decompensated cirrhosis who received FMT and SOC treatment for decompensated cirrhosis.
Group 2[Standard of care (SOC Group)]: Patients with DC who received only SOC treatment for decompensated cirrhosis.
SOC comprised of nutritional recommendation of a salt-restricted (<2 gm/day) and high-protein
diet (1.5-2 g/kg/day) diet with a targeted caloric intake of 35-40 kcal/kg/day. The patients underwent periodic nutrition counseling and reassessment at every visit. Anti hepatic encephalopathy measures (lactulose, rifaximin), intravenous albumin (as per standard recommendations), diuretics, beta-blockers, multivitamins, and calcium supplementation were continued as per indications and requirement. Any episode of suspected variceal bleed was managed with proton pump inhibitors, vasoconstrictors, and endoscopic intervention as indicated.
Stool Donor Selection:
Stool donor selection was done in a two-step process. Firstly, identified family members willing to become a stool donor were interviewed to assess history and risk factors for eligibility for being an FMT donor as per recommended guidelines.11 In brief, donors, were excluded if they had abdominal complaints, history of recent abdominal infections or been on antibiotics within the previous two months, had a history of chronic gastrointestinal diseases, history of luminal gastrointestinal surgery, history of any malignancy, autoimmune/atopic or neurological conditions, history of extensive travel history predisposing factors for potentially transmittable diseases, occasional or chronic alcohol intake or other substance abuse or had any state of primary or secondary immunosuppression. Once found suitable, the donor underwent detailed stool, blood, and urine tests to ensure that known transmissible diseases would not be passed along to recipients through FMT as per laid-out guidelines.11
Preparation of Donor Stool
Donors collected and submitted a fresh stool sample on the day of FMT after arriving at the hospital in sterile plastic collection containers. All personnel in stool specimen preparation wore personal protective equipment and performed the procedure in a pre-designated zone. All stool samples were obtained at least 6 hours before the procedure. Stool specimens with a weight of ∼30 g were taken as adequate. 100 mL of sterile normal saline was added to the stool sample and homogenized with an electronically operated blender for three cycles of thirty seconds each. The homogenous suspension was then filtered with filter paper and tea strainers 3-4 times until the filtrate was devoid of roughage.
Pre FMT Preparation
Participants randomized to the FMT group received pretreatment oral antibiotics (metronidazole 400 mg three-time daily, ciprofloxacin 500 mg twice daily, and amoxicillin 500 mg three times daily) for five days to reduce the host gut bacterial load and enable donor microbiome colonization.8 Lactulose and rifaximin were continued for all patients as per indication. All antibiotics were discontinued 12 hours before FMT to prevent modulation of administered FMT. Participants in the SOC group did not receive this pre-therapy antibiotic but otherwise had the same follow-ups post-randomization.
The FMT Procedure
In the FMT procedure, a 100 ml volume of strained and filtered stool was delivered through a nasojejunal (NJ) tube in two delivery sessions spaced 10 minutes apart in volumes of 50 ml each. The recipient patient was kept nil per oral for at least 4 hours before the stool instillation. The NJ tube was flushed with normal saline (50mL) after the stool instillation. The patients were allowed to consume a liquid diet two hours after the procedure. All patients continued SOC therapy, as advised in the management of DC.
Clinical and Laboratory Assessments
Clinical examination included a detailed evaluation of vital parameters, general physical examination, and a systemic examination. Laboratory investigations included a hemogram, renal and liver function tests, and a complete coagulogram. Liver disease severity was assessed using the Child–Turcotte–Pugh (CTP), MELD, and MELD-Sodium (MELD Na) scores. Blood samples from a peripheral vein were taken at baseline and days 7, 28, 90, and 180. Fasting ammonia Checker II (Daiichi Kagaku Co Ltd, Kyoto, Japan) using finger-prick capillary blood and measured at baseline, day 7, and day 28.
Assessment of Pro-inflammatory Cytokines
Cytokines (Interleukins IL-1 and IL6) were measured in plasma derived from patients using Human beta PicoKine ELISA kits (Boster Biological Technology) according to the manufacturer's protocol. The plate was read at 450 nm. Absorbance was converted to picograms per milliliter using a standard curve prepared with recombinant human IL1, IL6. Measures were taken at baseline and 28 days post-FMT
Follow Up
Follow-up of patients was done on days 7, 28, 90, and 180 in both groups. During follow-up, patients were evaluated for clinical parameters, routine biochemical monitoring, and inflammatory markers.
Adverse Events
All adverse events were recorded and graded according to Common Terminology Criteria for Adverse Events (CTCAE). Any event that resulted in death was life-threatening, required inpatient hospitalization, extended an ongoing hospital stay, or interfered substantially with normal life functions was considered a serious adverse event.
Statistical Analysis
The results are expressed as number (proportion) for categorical data, mean (95% Confidence Interval; CI or standard deviation; SD) for normally distributed numerical data, or median (range) for skewed numerical data. Comparisons between groups for numerical data were performed using student's t-test or the Mann–Whitney U test. For categorical data, the chi-square test or Fisher's exact test were applied. For intra-group comparisons, a repeated measures Analysis of Variance (RMANOVA) with a Greenhouse-Geisser correction was performed. A value of P < 0.05 (two-tailed) adjusted for multiple comparisons was taken as significant. The in-hospital survival curves were made by the Kaplan-Meier method and compared with the Log-Rank test. All statistical tests were done by Microsoft Excel and SPSS Software version 18 for Windows.
Sample size calculation
The only previous study which has looked at the impact of FMT on the survival of patients with liver disease, albeit in a population of severe alcoholic hepatitis, found a 75% survival in the FMT group and a 29% survival in the SOC group at 90 days.12. However, given the exploratory endpoints of this trial in patients with decompensated cirrhosis, a population which has previously not been looked at, as well as an unexpectedly low acceptance rate of FMT as a therapeutic modality in our population, we could enrol only 18 patients in each group in the pre-specified time period.
{kind=link}