2.1 Materials and Reagents
All DNA sequences were synthesized, labeled and purified by Sangon Biotechnology Inc. (Shanghai, China), and the sequences were shown in Table S1. Phosphate buffer solution (PBS), TM buffer, tris (hydroxymethyl) aminomethane (Tris), TE buffer, TBE buffer, and magnesium chloride (MgCl2) were purchased from Sangon Biotechnology Inc. One-Step RT-qPCR Kit and total RNA Extractor (Trizol) were purchased from Sangon Biotechnology Inc. Optiprep was purchased from Sigma-Aldrich Company Ltd. The human breast cancer cell line (MCF-7) and human cervical cancer cell line (HeLa) were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Dulbecco’s modified Eagle medium (DMEM; Gibco), Penicillin-streptomycin (PS), Fetal bovine serum (FBS; Gibco), 0.25% Trypsin-EDTA (1X), CellTracker™ Green CMFDA, CellTracker™ Orange CMFDA, Calcein (AM), Ethidium homodimer-1 (EthD-1), DAPI, and Hochest 33342 were purchased from Life Technologies. Alexa488-anti-EpCAM Rabbit monoclonal antibody (ab237395, Abcam) was obtained from Abcam Technology (Cambridge, UK). The SU-8-negative photoresist was purchased from MicroChem (MA, USA). Polydimethylsiloxane (Sylgard 184) and curing agent were obtained from Dow Corning (Shanghai, China). The film photomask was ordered from Gx Photomask Co., Ltd. (Shenzhen, China).
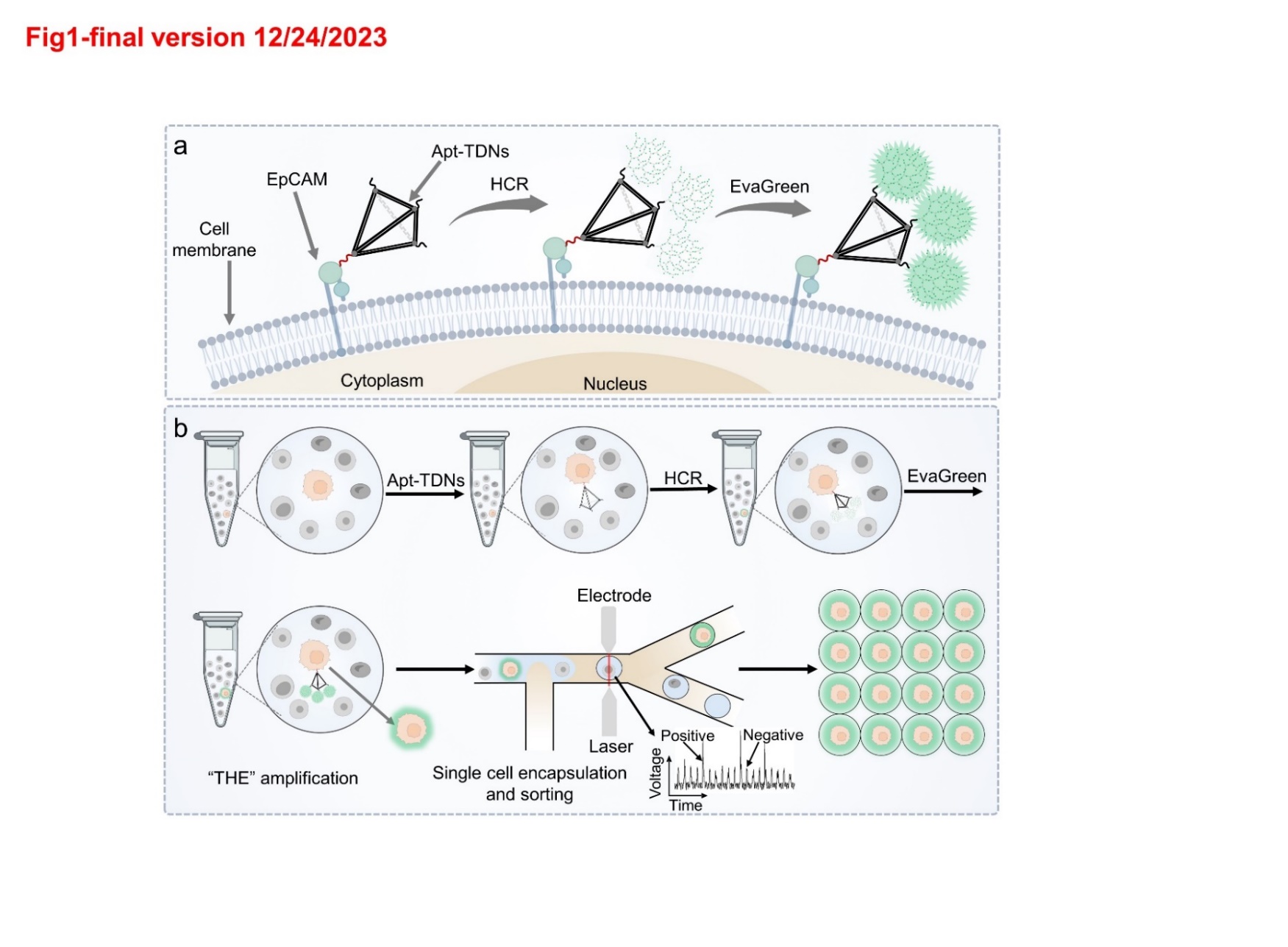
2.2 Preparation and characterization of the THE system
Apt-TDNs were synthesized according to the following protocol. Briefly, the aptamer strand and four base strands (tetra-A, tetra-B, tetra-C, tetra-D) were separately dissolved in TE buffer (10 mM Tris, 1 mM EDTA, pH = 8.0) to a final concentration of 10 µM. Each DNA strand with the same volume (5 µL) was mixed with TM buffer (25 µL, 20 mM Tris, 50 mM MgCl2, pH = 8.0). The resulting mixture was heated using a BioRad T100 thermal cycler with 95 ℃ for 10min, followed by cooling to 4 ℃ in 30 s. Apt-TDNs were identified by native polyacrylamide gel electrophoresis (PAGE). An 8% polyacrylamide gel solution (6 mL) was prepared with 30% acrylamide/bis-acrylamide solution (1.6 mL), MgCl2 (0.75 mL), 5 X TBE (1.2 mL), and Milli-Q water (2.45 mL). 60 µL ammonium persulfate (APS) and 6 µL 1,2-di- (dimethylamino) ethane (TEMED) were added into the polyacrylamide gel solution and mixed quickly for the preparation of PAGE gel. All the DNA samples were run at 100 V for 120 min. After electrophoresis, the gel was stained with GelRed for 15 min and visualized under UV illumination.
Apt-TDNs-dendrimers (HCR reaction-based dendritic structures) were synthesized based on the following protocol. Firstly, Substrate-A and Substrate-B were prepared separately by annealing mixtures of 3 µM F strand and 4.5 µM Q strand through a temperature cycle of 85°C for 5 min followed by cooling to ambient temperature with the rate of 1 ℃/s. Secondly, the annealed Substrates-A and Substrates-B were mixed with the corresponding 4.5 µM Assistants (Assistant-A and Assistant-B) separately and incubated for 20 min. Thirdly, these two resulting solutions were mixed with a ratio of 1:2 with the final concentrations of 0.5 µM Substrate-A, 0.75 µM Assistant-A, 1 µM Substrate-B, and 1.5 µM Assistant-B (H solution). Finally, Apt-TDNs were added into the H solution to form Apt-TDNs-dendrimers. The PAGE was employed to characterize the assembling of Apt-TDNs-dendrimers. Apt-TDNs at various concentrations (0.05, 0.1, 0.25, 0.5 µM) were mixed with the H solution to form Apt-TDNs-dendrimers with different sizes. Additionally, the H solution, 1 µM Apt-TDNs, 1 µM Substrate-A and 1 µM Substrate-B were also prepared for electrophoresis.
For atomic force microscopy (AFM) imaging of the Apt-TDNs-dendrimers, 10 µL 20 nM Apt-TDNs-dendrimers was diluted in TM buffer and subsequently filtered by a 0.22 µm membrane filter. The solution was then dropped in APTES pre-treated mica and adsorbed for 30 ~ 40 s. After this, the mica surface was rinsed four times with 50 µL water. Excess water was then removed using argon, and the mica was further dried under a vacuum for at least 20 min. AFM images were conducted using Dimension FastScan (Bruker, Germany). The identical protocol was applied for the AFM imaging Apt-TDNs.
For hydrodynamic size measurement of the Apt-TDNs-dendrimers, 200 µL 500 nM Apt-TDNs-dendrimers was diluted in water. 10 µL solution was carefully added into the bottom of a cuvette to avoid bubble formation. The dynamic light scattering (DLS) -based hydrodynamic size measurement was conducted by Zetasizer Nano ZS (Malvern, U.K.). The identical protocol was applied for the hydrodynamic size measurement of the Apt-TDNs.
The fluorescence intensity of the THE signal amplification system was characterized by a microplate reader. Firstly, H solution, 1X EvaGreen solution (E solution) and the mixture solution of H solution and E solution (H-E solution) were prepared, respectively. Secondly, 0.1 µM Apt-TDNs solution was added into the H solution, H-E solution respectively to trigger the HCR reaction for signal amplification. Lastly, the fluorescence intensity of all samples was measured by a microplate reader (Synergy H1, BioTek) with excitation and emission wavelengths at 485 and 525 nm, respectively.
2.3 Cell culture, imaging and fluorescence characterization
MCF-7 and Hela cells were cultured in the DMEM media supplemented with 10% FBS and 1% PS solution. All cells were maintained at 37°C in a 5% CO2 atmosphere. The cells were collected using 0.25% trypsin-EDTA when cultivated to 80–90% confluence in cell culture dishes, and the cell number was determined using a hemocytometer.
To demonstrate the signal amplification ability of the THE system on MCF-7 cells, experiments were divided into three groups: control group, HCR group and HCR-Eva group. For all experimental groups, the MCF-7 cells were diluted into 2 x 105 cells / mL. For the control group, the MCF-7 cells were centrifuged at 1000 rpm for 1 min, resuspended with 200 µL Apt-TDNs solution in binding buffer (5 mM MgCl2 in PBS, pH = 7.4). After incubation of 40 min on ice, the cells were washed with binding buffer. Then the MCF-7 cells were divided into two groups, one was for imaging by confocal microscopy (FV3000, Olympus), the other was centrifuged at 1000 rpm for 1 min and resuspended with 200 µL H solution. After incubation of 37 ℃ for 40 min, the cells were washed with binding buffer again. Then the MCF-7 cells were divided into two groups, one was for confocal imaging, the other was centrifuged at 1000 rpm for 1 min, resuspended with 200 µL E solution. After incubation of 37 ℃ for 20 min, the cells were washed with binding buffer, then imaged by confocal microscopy.
For the fluorescence intensity quantification, 104 MCF-7 cells from each experimental group were added into the wells of a microplate in 50 µL PBS solution. Fluorescence intensity was then measured with an excitation wavelength of 485 nm and an emission wavelength of 525 nm by a microplate reader.
To demonstrate the specific signal amplification ability of the THE system for MCF-7 cells, the Apt-TDNs solution, H solution and E solution were added into the MCF-7 cells or Hela cells solution separately, the following protocol for signal amplification of the THE system on MCF-7 cells was the same as above. Then the MCF-7 and Hela cells were imaged by confocal microscopy respectively.
For the cell viability characterization, the MCF-7 cells from all the experimental groups were stained with Calcein-AM/EthD-1 mixture (2 µM each) separately at 37°C for 20 min. Then the cells were imaged by confocal microscopy. The viable and dead cells were counted using ImageJ software. Cell viability was calculated as the ratio of viable cells to the total number of cells.
The MCF-7 cells from all the experimental group were also collected to perform reculture experiments. Briefly, the MCF-7 cells from each experimental group were seeded into a 24-well plate and cultured in DMEM media supplemented with 10% FBS and 1% PS solution at 37°C in a 5% CO2. The MCF-7 cells were imaged in 0 days and 2 days by confocal microscopy.
2.4 Microfluidic chip design and fabrication
The microfluidic chip was fabricated employing soft lithography techniques43, 44as the following steps: SU-8 3025 photoresist (MicroChem) was spin-coated at 1000 rpm onto a 3-inch silicon wafer to achieve a depth of ~ 60 µm for the first layer. Following a pre-bake at 95°C for 20 min, a photomask was positioned on the wafer, which was then exposed to 120 mW UV light (M365L2, Thorlabs) for 5 min. After a post-bake phase at 95°C for 10 min, a second coating of SU-8 3025 photoresist was applied at 1000 rpm and baked for 45 min. Then a second photomask was used, and the wafer was exposed to UV light for 5 min. Following a final bake at 95°C for 20 min, the wafer was developed in an SU-8 developer solution (MicroChem) for 15 min. The fabricated master was subsequently cleaned with isopropanol and ethanol, and then dried with a stream of nitrogen. A 10:1 (w:w) mixture of the PDMS precursor (SYLGARD 184, Dow Corning) and the curing agent was poured over the master, followed by an overnight curing process at 60°C. The PDMS slab was detached from the master, and a 0.7 mm hole puncher was employed to fabricate inlet/outlet ports. Oxygen plasma treatment was used to bond the PDMS slab to a glass slide. The chip was baked at 100°C for 30 min and 65°C overnight to enhance this bonding strength. Lastly, the channels in the chip were wetted by Aquapel (PPG Industries) to render the channel surfaces hydrophobic. Then the chip was baked at 60°C for 10 min before use.
2.5 Construction and optimization of the FADS
The designed FADS microfluidic chip integrated a primary fluidic component and an auxiliary component. The primary fluidic component comprised three inlets (cell, spacer, bias) and two outlets (waste, sorted). The cell inlet was used to input the cell solution. The spacer inlet and bias inlet were injected with a fluorinated oil containing 2% (v/v) PEG-PFPE surfactant (oil phase) separately. The waste outlet was connected to a suck pump and was used to collect the undesired droplets (empty droplets or droplets with un-target cells). The sorted outlet was used to collect the desired droplets (droplets with target cells). The auxiliary component comprised a moat, an electrode, and two optical fibers. The electrode and moat were injected with 4 M KCl solution. Two optical fiber channels were inserted with laser optical fiber and a PMT optical fiber separately. The electrode and moat were both filled with 4 M KCl solution. The dye in the droplet was excited by laser fiber with a 473 nm laser, and a PMT fiber was used to detect the fluorescence signal in real time. The FADS microfluidic chip was placed on a sorting machine (CytoSpark droplet sorting system, Zhejiang Dapu Biotechnology Co., Ltd).
To evaluate the practicability of the FADS system, the experiments were classified into control group, antibody group, HCR group and HCR-Eva group. For the antibody group, the MCF-7 cells were incubated with 5 µg/µL EpCAM antibody for 40 min on ice, then the cells were washed with binding buffer. For the other three groups, the signal amplification of the THE system on MCF-7 cells followed the protocol in “cell culture, imaging and fluorescence characterization” part. The MCF-7 Cells from each experimental group were separately introduced into the FADS microfluidic chip. The flow rate of cell inlet, spacer oil inlet, bias oil inlet and waste outlet were set as 100 µL/h, 400 µL/h, 500 µL/h and 500 µL/h, respectively. After the flow rate was stable, the signals of droplets across each group were collected.
2.6 MCF-7 cells sorting by the THE-FADS in PBS solution
MCF-7 cells and Hela cells were stained with 5 µg/mL DAPI and 5 µM Cell Tracker Orange for 15 min respectively, then mixed together with the ratio of 1:1. After that, the Apt-TDNs solution, H solution and E solution were added into the cell mixture sequentially. The signal amplification of the THE system on MCF-7 cells in the cell mixture followed the protocol in “cell culture, imaging and fluorescence characterization” part. Then the cell mixture was centrifuged at 1000 rpm for 1 minute. After the supernatant was discarded. The cell mixture was resuspended in OptiPrep-PBS (20% OptiPrep in PBS with 0.02% BSA) and loaded into a 1ml syringe connected to the cell inlet of the FADS microfluidic chip with the flow rate of 100 µL/h. The spacer inlet and bias inlet were filled with HFE-7500 fluorinated oil containing 2% (v/v) PEG-PFPE surfactant by a 2 mL syringe, with a flow rate of 400 µL/h and 500 µL/h, respectively. The waste outlet was constantly sucked by a connected 2 mL syringe with the flow rate of − 500 µL/h. A 2 V PMT voltage threshold was set to perform sorting. The sorted droplets with MCF-7 cells were collected in a 1.5 mL Eppendorf tube and imaged by confocal microscopy.
To test the sorting purity of the THE-FADS system, the MCF-7 cells were diluted into varying numbers (20, 200, 1000, 2000) in a 200 µL OptiPrep-PBS solution, the Hela cells were prepared with the concentration of 2 x 105 cells / mL. The protocol of MCF-7 cells and Hela cells for staining, mixing together with signal amplification of the THE system on MCF-7 cells was the same as above except that the MCF-7 cells with varying numbers mixed with Hela cells. The sorting purity was defined as the ratio of sorted MCF-7 cells to the total number of sorted cells.
To test the sorting efficiency of the THE-FADS system, the MCF-7 cells were diluted into varying numbers (20, 200, 1000, 2000) in a 200 µL OptiPrep-PBS solution. After that, the Apt-TDNs solution, H solution and E solution were added into the MCF-7 cells sequentially. The signal amplification process of THE system on MCF-7 cells followed the protocol in “cell culture, imaging and fluorescence characterization” part. Then the MCF-7 cell solution was injected into the FADS microfluidic chip for sorting. The sorting efficiency was defined as the ratio of sorted MCF-7 cells to the total number of spiked MCF-7 cells.
For FACS-based MCF-7 cells sorting, the MCF-7 and Hela cells were mixed with the ratio of 1:100 and incubated with 5 µg/µL EpCAM antibody for 40 min on ice. Then the cell mixture was washed with binding buffer. The cell mixture was imported into a flow cytometer (FACS Aria III, BD Biosciences) to sort MCF-7 cells.
To compare the cell viability of MCF-7 cells obtained by the THE-FADS system and the antibody-based FACS system, 4,000 MCF-7 cells from each sorting system were recultured. After three days, the MCF-7 cells were imaged by confocal microscopy.
2.7 MCF-7 cells sorting by the THE-FADS in the mimic serum samples and downstream analysis
The staining and mixing of MCF-7 cells and Hela cells, signal amplification of MCF-7 cells by the THE system was the same as that in PBS solution except that the ratio of MCF-7 cells and Hela cells in mixture solution was 1:100. The sorting procedure was the same as that in PBS solution except that OptiPrep-FBS (20% OptiPrep in FBS) was used to replace PBS. After obtaining the MCF-7 cell-laden droplets, the cells were recovered by adding 100 µL DMEM media supplemented with 10% FBS, followed by 100 µl 1H,1H,2H,2H-perfluoro-1-octanol. The mixture was then gently mixed and centrifuged at 700g for 1 min to favor complete phase separation. The bottom oil was discarded, and the MCF-7 cells were obtained.
For downstream molecular analysis, the MCF-7 cells sorted by the THE-FADS in mimic serum samples were recultured and imaged by confocal microscopy. After the MCF-7 cells were cultivated to 80–90% confluence, the cells were digested with trypsin. Total RNA was extracted from the recultured MCF-7 cells using Trizol according to the manufacturer’s instructions. The normal cultured MCF-7 cells were set as the control. Extracted RNA was used in a one-step RT-qPCR reaction with Taq DNA Polymerase to amplify CK19 and EGFR genes. The PCR procedures were as follows: 50°C for 5 min, 95°C for 3 min, 40 cycles of 95°C for 10 seconds and 60°C for 30 seconds.
{kind=link}