Animals
All mice were bred on a C57BL/6 background and raised in a specific pathogen-free (SPF) environment. Wild-type mice were purchased from GemPharmatech (Guangdong, China). The Hmgcs2flox mouse line was generated using CRISPR/Cas9 technology by Gempharmatech (Nanjing, Jiangsu, China). Male mice were identified by PCR performed on DNA isolated from the tail. All mice were housed in the Sun Yat-sen University Animal Center under specific conditions of constant temperature (24 ± 1°C), relative humidity (50–60%), and a 12 h light/12 h dark cycle. They were provided ad libitum access to food and water throughout the study. The animal use protocol has been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC), Sun Yat-Sen University (Approval No. SYSU-IACUC-2022-001335). Strict adherence to animal welfare and ethical considerations was ensured throughout the study.
Sample Preparation for Single Cell RNA Sequencing
2-month-old and 24-month-old mice were anesthetized with Avertin (250 mg/kg) by i.p. injection. After dissection, the testes were immediately placed in ice-cold saline. After fully washing the testes with PBS, the tunica albuginea was carefully removed. The testes were mechanically minced into pieces using microscopic forceps and digested with accutase (Invitrogen, USA) at 37°C for 5 minutes. The accutase activity was halted by adding DMEM/F12 supplemented with 10% fetal bovine serum (Gibco, USA), and the samples were rinsed twice with PBS. The obtained cell suspension was filtered through a 45 µm strainer and centrifuged. At last, cells were resuspended in phosphate-buffered saline (PBS) containing 0.1% BSA (Thermo Fisher Scientific, USA) at a concentration of ~ 1,000 cells/µL for single-cell sequencing.
Single Cell RNA-seq Performance, Library Preparation and Sequencing
The BD Rhapsody system was used to capture the transcriptomic information. Single-cell capture was achieved by randomly distributing a single-cell suspension across microwells using a limited dilution approach. Beads with oligonucleotide barcodes were added in excess to ensure that a bead paired with a cell in each microwell. Cell-lysis buffer was added to allow poly-adenylated RNA molecules to hybridize to the beads. The beads were collected into a single tube for reverse transcription. Whole transcriptome libraries were prepared using the BD Rhapsody single-cell whole-transcriptome amplification workflow. Sequencing libraries were quantified using a High Sensitivity DNA chip (Agilent, USA) on a Bioanalyzer 2200 and the Qubit High Sensitivity DNA assay (Thermo Fisher Scientific, USA). The library for each sample was sequenced using Illumina sequencer (Illumina, CA).
Ketone bodies measurement
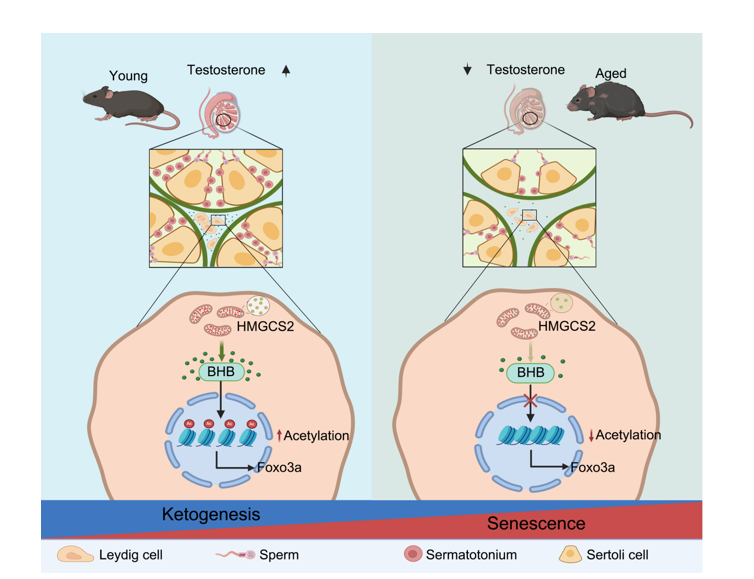
The concentration of BHB in the blood was quantitatively measured using a blood ketone meter (Abbott, USA). Blood samples were collected from the tail vein of mice using standard procedures. A small incision was made using a sterile scalpel and the tip of the strip was then brought into contact with a blood droplet from the tail vein. The strip drew the whole blood into the blood channel via capillary action. The meter starts measuring the amount of BHB when the channel is completely filled with blood.
To detect the concentration of AcAc in the blood, blood was collected from the mandibular vein of the mice and left to clot for 1 hour. The samples were then centrifuged at 5000g for 10 minutes, and the supernatant serum was collected for subsequent analyses. Supernatant were tested according to the protocol provided by the colorimetric kit (Abcam, USA) manufacturer.
To detect the concentration of ketone bodies in the testes, colorimetric kits were used to analyze AcAc (Abcam, USA) and BHB (Cayman, USA). Testes were collected from the mouse and placed in 1 ml of saline with two 4 mm beads (Easybio, China). A high-throughput tissue lyser was used to grind the testicular tissue at a frequency of 50 Hz for 5 minutes. Subsequently, the sample was centrifuged at 10,000 g for 3 minutes, and 100 µl of the supernatant was collected and diluted tenfold with saline for ketone bodies detection according to the protocol provided by the colorimetric kit manufacturer.
To detect intracellular ketone body concentration, colorimetric kits were used to analyze AcAc (Abcam, USA) and BHB (Cayman, USA). The cell culture medium was discarded, the cells were washed three times with saline, and then 1 ml of 80% methanol solution (pre-cooled at -20°C for 6 hours) was added. After lysis, a cell scraper (NEST, China) was used to transfer the mixture to a 1.5 ml centrifuge tube. The mixture was vortexed for 5 minutes, then centrifuged at 13,000g for 10 minutes at 4°C, and the supernatant was collected for subsequent detection. Supernatant were tested according to the protocol provided by the colorimetric kit manufacturer.
Isolation of LCs: Isolation of LCs from wild-type mice was performed as previously described by our group54. In brief, the tunica albuginea was carefully removed and then the testes were mechanically minced and enzymatically dissociated using 200 µg/mL DNase I (Gibco, USA) and 1 mg/mL Type IV Collagenase (Gibco, USA) in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12; 1:1, Gibco, USA) at 37°C for 15 minutes with gentle agitation (100 cycles/min). The collagenase activity was halted by adding DMEM/F12 supplemented with 10% fetal bovine serum (Gibco, USA), and the samples were rinsed twice with PBS. The resulting cell suspension was filtered through a 45 µm strainer to obtain a single-cell suspension. LCs were subsequently sorted using flow cytometry (Beckman Coulter, USA). For Isolation of LCs from AAV infected testes, prepare the single-cell suspension according to the above method. GFP + or mCHERRY+ LCs were subsequently sorted using flow cytometry (Beckman Coulter, USA).
Production of lentivirus
HEK293T cells were cultured in DMEM (Corning, USA) supplemented with 10% FBS, 1% Penicillin-Streptomycin (Gibco, USA), and 1% GlutaMAX (Gibco, USA). To package lentivirus for the construction of a HEK-293 reporter cell line, HEK293T cells were seeded in a T75 flask (NEST, China). When cells reached 90% confluency, 4.5 µg of lentiviral transfer plasmid, 3 µg of psPAX, and 1.5 µg of pMD2.G were co-transfected using Lipo8000™ Transfection Reagent (Beyotime, China). Twelve hours after transfection, the media was replaced. At 48 and 72 hours post-transfection, the supernatant was collected. The collected supernatant was then filtered through a 0.45 µm sterile filter (Millipore, USA). Subsequently, the lentivirus was centrifuged at 50,000 g for 90 minutes, resuspended in 0.5 mL PBS, and stored at − 80°C.
Construction of gene knockdown cell line
MLTC-1 cells were cultured in 1640 (Corning, USA) supplemented with 10% FBS (Gibco, USA), 1% Penicillin-Streptomycin (Gibco, USA), and 1% GlutaMAX (Gibco, USA). Sh-RNA sequences were cloned into pLKO.1-copGFP-PURO plasmids by Singke Biotech. The plasmid was used for the production of lentiviral vectors. At 70% confluence, the cells were transfected with lentivirus containing shRNA sequences in the presence of 8 µg/mL polybrene. Four days post-transduction, the cells were digested with 0.25% trypsin at 37°C for 3 minutes and then filtered through a 45 µm strainer to obtain a single-cell suspension. GFP+ cells were subsequently sorted using flow cytometry (Beckman Coulter, USA) for further experiments.
RNA extraction, cDNA synthesis, and quantitative RT-PCR
Total RNA was extracted from testes or cells using the RNeasy Mini Kit (Yibin, China) according to the manufacturer’s protocol. RNA purity was assessed by measuring the 260/280 ratio (1.87–1.93) and the 260/230 ratio (2.18–2.34) using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, USA). Reverse transcription was performed using the NovoScript® 1st Strand cDNA Synthesis Kit (Novoprotein, China). Quantitative RT-PCR was carried out using SYBR PCR Master Mix (Roche, CH) and a LightCycler 480 Detection System (Roche, CH). Primer validation was performed by generating a melting curve to confirm a single peak, thereby ruling out the possibility of non-specific product or primer dimer formation. β-actin served as an internal control, and target gene expression levels were calculated using the ΔCt method and expressed relative to β-actin.
Induce cell premature senescence and treatment with AcAc or BHB
To induce premature senescence, H2O2 (Hengjian, China) was added to the medium at a concentration of 100 µM. The subsequent steps are detailed in Fig. 5A. In brief, cells were treated with hydrogen peroxide for 48 hours. After adding hydrogen peroxide for 24 hours, BHB or AcAc was added to the culture medium. All groups were analyzed on the forth day. Doxorubicin (MCE, USA) was dissolved in DMSO, stored at -20°C, and then added to the culture medium at a concentration of 150 nM. The subsequent steps are detailed in Fig S4A. In brief, the cells were treated with doxorubicin twice, with a 24-hour interval between treatments. After adding doxorubicin for 24 hours, BHB or AcAc was added to the culture medium. All groups were analyzed on the fifth day. For inducing premature senescence by radiation, cells were exposed to 8 Gy of ionizing radiation using an RS 2000 X-ray Biological Irradiator (Rad Source, USA) at a dose rate of 1.185 Gy/min. The subsequent steps are detailed in Fig S4E. In brief, three days after the cells were irradiated, BHB or AcAc was added to the culture medium. Following the addition, the cells were cultured under standard conditions. 3-Hydroxybutyric acid sodium (MCE, USA) and Lithium acetoacetate (Sigma, USA) were both dissolved in saline, stored at -80°C, added to the culture medium at a concentration of 5 mM.
Chip-qPCR
A ChIP assay kit (CST, USA) was employed to assess the binding of H3K9ac to the promoters of Foxo3a. MLTC-1 cells were seeded in a T75 flask (NEST, China). Following treatment, cells at 70% confluence were fixed with 1% formaldehyde on ice to cross-link the proteins bound to the chromatin DNA. After washing, the chromatin DNA was enzymatically sheared to produce DNA fragments ranging from 200 to 1,000 bp in size. Subsequently, DNA recovery was performed according to the manufacturer's protocol. The DNA recovered from the reverse cross-linking was utilized for qPCR analysis. An equal amount of sheared DNA without antibody precipitation was processed for reverse cross-linking and utilized as an input control.
Western blot
For protein extraction from cultured cells, cells were washed three times with ice-cold PBS. Subsequently, the cells were lysed directly in RIPA buffer (Beyotime, China) for 30 min, and subjected to centrifugation at 13000 g for 10 min at 4°C. Each supernatant was recovered as a total cell lysate. For protein extraction from tissue, testes were grinded in liquid nitrogen and subsequently lysed with RIPA Buffer (Beyotime, China). The lysates were placed on ice for 30 minutes and then centrifuged at 12,000g for 15 minutes at 4°C. Supernatants containing proteins were collected, and protein concentrations were determined using Pierce™ BCA Protein Assay Kits (Thermo Fisher, USA). The lysates were resolved by SDS polyacrylamide gel electrophoresis and then transferred onto 0.45 µm pore-sized polyvinylidene fluoride membranes (Millipore, USA). Subsequently, the membranes were blocked with 5% milk (Phygene, China), incubated with primary antibodies overnight at 4°C, washed with tris-buffered saline Tween-20 (TBST), and subsequently incubated with HRP-conjugated secondary antibodies for 1 hour at room temperature. Finally, the signals were visualized using an ECL substrate (NCM, China). Immunoreactivity was semi-quantitatively detected by ChemiDoc Imaging Systems (Bio-Rad, USA)
Immunofluorescence staining
For immunofluorescence staining of sections, the tissues were fixed in 4% paraformaldehyde (PFA; Phygene, China) at 4°C for 4 hours, dehydrated with 30% sucrose (Sangon Biotech, China), and sectioned at a thickness of 10 µm. The sections underwent antigen retrieval using Citrate Antigen Retrieval Solution (Beyotime, China), followed by permeabilization with 0.2% Triton X-100 (Sigma, USA) for 15 minutes. After blocking with 2% BSA (Sigma, USA) and 10% goat serum (Boster, China) in PBS for 45 minutes at room temperature, sections were incubated overnight with primary antibodies. Subsequently, sections were washed with PBS five times, incubated with appropriate secondary antibodies for 60 minutes at room temperature, and counterstained with DAPI (Gibco) for 5 minutes. Images were captured using an LSM800 confocal microscope (Zeiss, Germany) or a Nikon C2 confocal microscope (Nikon, Japan). For immunofluorescence staining of cells, the Cells were seeded in a 24-well plate and fixed in 4% PFA (Phygene, China) for 15 minutes. Fixed cells were permeabilized with PBS containing 0.2% Triton X-100 for 15 minutes at room temperature. Non-specific antibody binding was blocked with 2% BSA and 10% goat serum in PBS for 45 minutes at room temperature, followed by overnight incubation with relevant primary antibodies at 4°C. Next, cells were washed three times with PBS and incubated with appropriate secondary antibodies for 45 minutes at room temperature. Finally, the nuclei were counterstained with DAPI for 5 minutes, and images were captured using an LSM800 confocal microscope (Zeiss, Germany) or a Nikon C2 confocal microscope (Nikon, Japan).
H&E staining
The testes were collected, fixed overnight in Bouin’s solution (Sigma, USA), dehydrated in 75% ethanol, embedded in paraffin, and then sectioned into 4 µm-thick slices. Subsequently, the paraffin-embedded sections were deparaffinized using xylene and then gradually rehydrated with a series of ethanol concentrations. For histological analysis, the prepared sections were stained with haematoxylin and eosin (H&E). The stained sections were examined using KF-PRO-120-H1 slide scanner (KFBIO, China).
SA-β-gal staining
According to the manufacturer’s protocol for the senescence-associated β-galactosidase kit (Beyotime, China), cells seeded in 12-well plates were washed with PBS and subsequently fixed in β-galactosidase staining fixative (Beyotime, China) for 15 minutes. After washing, the cells were incubated overnight with the working solution at 37°C. Senescent cells were observed and counted under an DMi8 microscope (Leica, Germany) from three random fields of view. For SA-β-gal staining of tissue sections, the O.C.T (optimal cutting temperature compound)-embedded testicular tissues were cryosectioned at a thickness of 10 µm, and stored at − 80°C. Before SA-β-Gal staining, sections were thawed at RT and rinsed in PBS for 1 min, fixed in in β-galactosidase staining fixative (Beyotime, China) at RT for 15 min. The sections were then stained according to the manufacturer’s protocol for 6 hours. After staining, the sections were retrieved using Citrate Antigen Retrieval Solution (Beyotime, China), permeabilized with 0.2% Triton X-100 (Sigma, USA) for 15 minutes, blocked with 2% BSA (Sigma, USA) and 10% goat serum (Boster, China) in PBS for 45 minutes at room temperature, and then subsequently incubated overnight with primary antibodies. Following incubation, the sections were washed five times with PBS, incubated with appropriate secondary antibodies for 60 minutes at room temperature, and images were captured using DMi8 microscope (Leica, Germany).
Computer-aided semen analysis
Semen samples were analysed as previously reported30. In brief, two cauda epididymides were harvested from each mouse, incised with micro scissors, and then incubated in 1 mL DMEM/F12 containing 0.5% BSA for 15 min at 37°C to allow for sperm release. The tissue was removed, and sperm samples were analysed using a Hamilton Thorne's Ceros II system. At least six fields were assessed for each sample, and the sperm concentration and percentages of motile and progressively motile spermatozoa were determined.
Sex hormone assays
Sex hormone concentrations were assayed as previously reported by our group55. In brief, blood was collected from the mandibular vein of the mice and left to clot for 1 hour. The samples were then centrifuged at 5000g for 10 minutes, and the supernatant serum was collected for subsequent analyses. For intratesticular testosterone concentration detection, testes were extracted from the mouse and placed in 1 ml of saline with two 4 mm beads (Easybio, China). A high-throughput tissue crusher was used to crush the testicular tissue at a frequency of 50 Hz for 5 minutes. Subsequently, the sample was centrifuged at 10,000 g for 3 minutes, and 100 µl of the supernatant was collected and diluted tenfold with physiological saline. Serum and testes samples were collected at the indicated time points and stored at -80°C until analysis. Testosterone levels were measured using a chemiluminescent immunoassay (CLIA) (KingMed Diagnostics Group Co, Ltd, China). The minimum detectable dose of testosterone is 0.01 ng/mL. The concentration of serum LH and FSH was calculated based on the instructions provided by the manufacturer using an Elisa kit (Cloud-clone, China). The concentration of serum Insl3 was calculated based on the instructions provided by the manufacturer using an Elisa kit (Phoenix, USA). To detect the ability of cells to secrete progesterone, MLTC-1 cells were washed three times with PBS, and then fresh 1640 medium containing 10% FBS and 1% GlutaMAX was added. After 4 hours, the culture supernatant was collected for subsequent detection, and the cells were digested with 0.25% trypsin for cell counting using a computer-assisted system (Nexcelom, USA).
Gene delivery in animal models
For Hmgcs2 overexpression, the full-length mouse Hmgcs2 cDNA was cloned into the AAV-CAG vector. Wild-type aged mice (15-month-old) were intratesticularly injected with AAVDJ-Hmgcs2 for Hmgcs2 overexpression. For conditional knockout (CKO), the full-length Cre cDNA was inserted into the AAV-CAG vector. Eight-week-old male Hmgcs2flox/flox mice were intratesticularly injected with AAVDJ-Cre for conditional knockout. Following sequencing-based verification, the constructed vector was custom-packaged, purified, and titrated by Packgene Bioscience (Guangzhou, China). The viruses were stored at -80°C until use. For gene delivery, mice were anesthetized with Avertin (250 mg/kg) via intraperitoneal injection. The injection site was sterilized with ethanol and a topical application of povidone-iodine. The mouse testes were fixed through the scrotum with fingers, and AAV particles were injected into the testes using an insulin syringe (20 µL per testis; 2×10^9 genome copies per testis). After injection, the needle was left inside the tissue for 30 seconds to allow the virus to spread evenly within the testis.
BHB supplementation in aged mice
Wild-type aged mice (18-months-old) received sodium/potassium salt supplementation in their drinking water. The KetoForce solution is a BHB salt with 1.6g sodium and potassium per 11.7g BHB. 10 ml BHB salt formulation (KetoForce) was administered in the 290ml drink water for BHB supplementation. Add 3.2g of sodium chloride and 2.4g of potassium chloride to 250ml of water for the control group to drink. The experimental group and the control group were supplemented with equivalent sodium and potassium.
Statistical analysis
All data were subjected to statistical analysis using IBM SPSS Statistics version 25.0 software (IBM SPSS Statistics, Armonk, NY, USA) and the results were visualized using GraphPad Prism 9 software (GraphPad Software, La Jolla, CA, USA). The bioinformatics data were statistically analysed using was determined by Mann-Whitney U test and p or adjusted p values were indicated in each figure. For experiments except RNA-seq data, statistical differences between samples were assessed with Student’s t-tests, one-way analysis of variance (ANOVA) or two-way ANOVA. Differences were considered significant when p < 0.05 (*p < 0.05, **p < 0.01 and ***p < 0.001).
{kind=link}