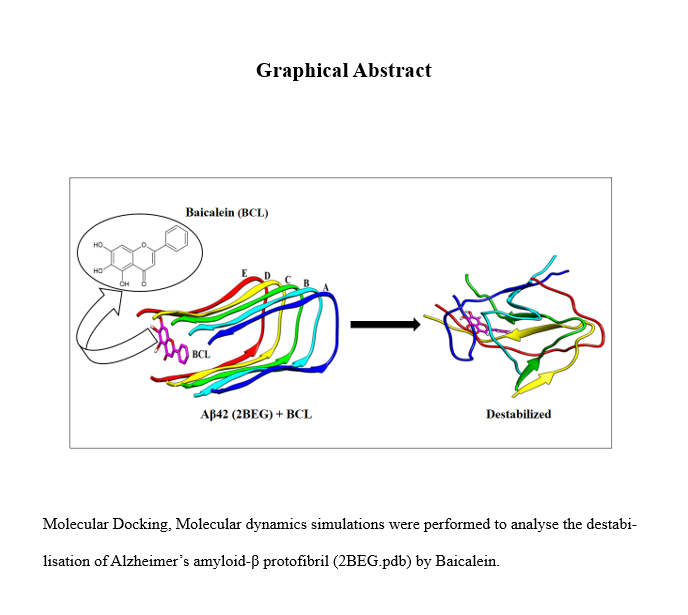
Molecular docking, evaluating Aβ-BCL binding mode
The BCL molecule was first docked to the three-dimensional structure of Aβ42 oligomer (PDB ID: 2BEG), using CB-Dock2, a blind docking software. Without prior knowledge of the target pockets, blind docking protocol scans the entire surface of the receptor protein for possible binding sites. We chose the highest-ranked pose with a binding energy of -6.9 kcal/mol. The docked complex was visualised using UCSF-Chimera, as shown in Fig. 2. The high interaction energy signifies that BCL is an excellent binder to the Aβ protofibril, and can potentially bring conformational changes in the protein.
The BCL molecule binds to the outer U-shaped cavity of the protofibril, formed by the β1 and β2 strands. In the Aβ42 model considered in our study, the L17 residue points outward while F19 is directed inwards, creating a large pocket surrounded by the terminal residues: L17, V18, F19, G38, V39, and V40. The other protein cavities are buried deep inside the fibril. Thus, the larger volume of the outer cavity and its proximity to the solution environment make it a more favourable binding site.
The analysis of the docked structure reveals that besides forming hydrophobic interaction, the BCL molecule also forms a hydrogen bond with the V39 residue of chain E and pi-stacking with the F19 residue of chain D, as shown in the protein-ligand interaction diagram (Fig. 2). The residues in contact with the ligand were also tabulated (Table 1) and depicted in Supplementary Fig. S1. The interaction of the BCL molecule with the hydrophobic residues of protofilament enables its access to the hydrophobic cavity. The findings were consistent with previous studies where compounds docked in the outermost cavity were found to destabilise the Aβ oligomer[62, 63].
Table 1
|
Docked Complex
|
Vina Score
(kcal/mol)
|
Residue in Contact
|
|
Aβ-BCL
|
-6.9
|
Chain B: PHE 19
Chain C: LEU 17, VAL 18, PHE 19
Chain D: LEU 17, VAL 18, PHE 19, VAL 39, VAL 40
Chain E: LEU 17, VAL 18, PHE 19, GLY 37, GLY 38, VAL 39, VAL 40
|
Validation of computational data with experimental results
Before assessing the impact of the BCL molecule on the Aβ42 protofibril, we validated the conformational ensemble generated through molecular dynamics (MD) simulations against experimental structures. This involved calculating NMR chemical shift values for Cα and Cβ atoms using SHIFTX2[64], which were then compared with experimental data[65]. The average computational Cα and Cβ chemical shifts (δ sim) showed a robust correlation of 0.94 and 0.99, respectively, with the experimental chemical shifts (δ exp), as depicted in supplementary Fig. S4. The comparison demonstrated strong agreement between experimental and computational data for the Aβ42 protofibril, consistent with previously reported results[66], affirming the reliability of our MD simulation data. Additionally, three-bond J–coupling (3JNH−Hα) constants were also analysed. These constants reflect the three-bond coupling interaction between HN and Hα protons, providing further insights into peptide conformations. The three-bond J-coupling constants, shown in Supplementary Fig. S5, were derived via the Karplus equation[67] using the Vuister and Bax parameters[68]. The J coupling constants of the Aβ42 protofibril show significant agreement between experimental and simulated outcomes.
MD simulation of Aβ protofilament in the presence of BCL
The simulations were carried out for 300 ns for all the systems following the protocols discussed in the method section. The final configurational snapshots of the systems at 300 ns were taken. We can observe from Fig. 3a that the protofibril in the control system was reasonably stable and showed only slight twisting of the peptide chains at the edges, and a marginal outward movement of chain A. The twists of the β strands increase protofibril's stability further by increasing the side chain packing[69]. However, the peptide chains has been highly destabilised by the introduction of BCL molecule, as shown in Fig. 3b. The disorganised structure of the protofibril in the Aβ-BCL systems prompted us to study the disruptive effect of the ligand further. Hence, other parameters were evaluated, as discussed below.
Destabilisation of the Aβ oligomer
In the presence of the BCL molecule, the Aβ oligomer was destabilised. The degree of destabilisation was measured by using various global stability parameters, such as, Cα-RMSD, Rg and SASA. The average values of these parameters across the 300 ns trajectory were plotted for both the Aβ-BCL and Aβ-Water systems and were compared quantitatively.
The Cα-RMSD of the protein was used to assess the structural stability of the protofibrils. It was found that the average RMSD value was increased to 1.17 ± 0.13 nm in the presence of the BCL molecule as compared to the 0.92 ± 0.09 nm of the control (viz., Aβ-Water) system. The same can also be observed from the average Cα-RMSD plot for the protein (Fig. 4a), where the curve for the Aβ-BCL is shifted upward towards a higher RMSD, unlike the BCL-free system. A similar trend in the RMSD plot was observed in previous destabilisation studies of the oligomer[70, 71]. The twisting of chains A and E has contributed the most to the RMSD of the control systems. This can be attributed to the limited contact that peripheral chains have with other chains, leading to a tendency to deform in order to maximise contact[72]. However, the overall U-shaped motif formed by the β1 and β2 strands was preserved.
The RMSD values from the β1, β2, and turn regions were calculated to expound more information about the most affected areas of the protofibril. It was found that all the regions showed an increased RMSD in the presence of the BCL molecule. The average RMSD for β1, β2 and turn region has been increased from 0.66 ± 0.10 nm, 1.07 ± 0.08 nm and 0.96 ± 0.16 nm in the Aβ-Water system to 1.09 ± 0.20 nm, 1.18 ± 0.09 nm and 1.09 ± 0.13 nm in the Aβ-BCL system, respectively. The difference in the RMSD value is more pronounced in the β1 region, as can be observed from Fig. 4b. The average RMSD values of these regions were plotted with the associated standard deviation represented as error bars. The increased RMSD suggests that the BCL molecule caused significant conformational changes in the protofibril.
The Radius of gyration (Rg) measures the compactness of a protein structure. Therefore, we measured the Rg values to evaluate whether the BCL molecule could increase the mobility of the chains, and hence decreasing the rigidity of the fibril structure Our results align well with the RMSD analysis. The average Rg value of the protein was found to be 1.47 ± 0.02 nm and 1.52 ± 0.02 nm for Aβ-Water and Aβ-BCL systems, respectively (Fig. 5a). A comparable increase in Rg values was also observed in a previous study focusing on the destabilization of Aβ protofibrils using Arginine-containing short peptides[73].
We have measured the solvent-accessible surface area (SASA) of the protofibril (Fig. 5b) to estimate how tightly the chains are packed. The non-polar residues are packed together in the hydrophobic core during protein folding, and they are essential for maintaining protein structure and stability. The average SASA value of the fibril has been increased significantly from 81.09 ± 2.19 nm2 to 86.61 ± 2.21 nm2 in the presence of the BCL, indicating loosening of the fibrillar structure. Thus, increasing SASA suggests that the BCL molecule is capable in disrupting the hydrophobic packing of the protofibril by increasing its exposure to water. The increase in both the average Rg and SASA values in the Aβ-BCL system indicates that the perturbation has been escalated in the presence of BCL, proving its potency as a destabiliser.
Changes in the secondary structure
Aβ monomers with initial β-sheet structures self-assemble into oligomers and aggregate into toxic fibril via a primary nucleation mechanism [74]. The formation of β-sheet features is a critical early step in Aβ amyloidogenesis and neurotoxicity[6]. Hence, evaluating the change in secondary structure of the protofibril would be valuable in comprehending the anti-aggregation effect of the flavonoid. We assessed the impact of the BCL molecule on the β-sheet structure content of Aβ peptide in the pentameric protofibril state using the DSSP (dictionary of secondary structure of proteins) method.
Figure 6a illustrates that the Aβ protein had a high β-sheet content in the water system. Upon interaction with the BCL molecule, there was a noticeable increase in coil structures at the expense of β-sheet content over the 300 ns trajectory, as shown in Fig. 6b.
It is important to note that converting the β-sheets into coil or α-helices is a crucial step in impeding primary nucleation, and preventing the amyloid fibrillation process. Although the Aβ-BCL system preserves the β-strand to a certain extent, there were still noticeable alterations compared to the Aβ-Water system. There was particularly an increase in coil content and a decrease in β-sheet content, as shown in Table 2. The β-sheet content has been decreased from 37–30%, while the coil content has been increased from 39–46% in the system with the BCL molecule. The present findings are consistent with previous studies that have reported a reduction of β-sheet content of Aβ fibril resulting in its destabilisation upon exposure to D744, a fluorinated derivative of curcumin[66], and norepinephrine[75]
Table 2
Percentage of different secondary structure contents of Aβ-Water, and Aβ-BCL systems during 300 ns simulation
|
System
|
% β-Sheet
|
% Coil
|
% Bend/Turn/Helix
|
|
Aβ - Water
|
37
|
39
|
24
|
|
Aβ - BCL
|
30
|
46
|
24
|
Binding mode of BCL molecule
The MM-PBSA method was integrated with MD simulation to investigate the binding affinity of the BCL molecule with the protofibril chain and to calculate the energetic contribution of various interactions. For the MM-PBSA analysis, the last 10 ns of the trajectory was considered with ∆t = 10 ps.It was observed that both the non-bonded van der Waal (vdW) and electrostatic interactions favoured ligand binding (Table 3). However, the vdW interaction was almost ~ 10-fold more dominant than the electrostatic interaction. The contribution of the vdW energy (-146.64 ± 8.52 kJ/mol) dominates over all the other energy terms, contributing the most towards the negative ΔGbinding of 76.59 ± 10.23 kJ/mol. The non-polar solvation energy (-15.75 ± 0.83 kJ/mol), is also favourable for the formation of the complex. However, the same cannot be said for the polar component of solvation energy.
Table 3
Binding free energy between Aβ protofibril and BCL molecule
|
Energy terms
|
Aβ-BCL (kJ/mol)
|
|
ΔEvdW
|
-146.64 ± 8.52
|
|
ΔEelec
|
-15.49 ± 5.90
|
|
ΔGps
|
+ 101.30 ± 8.56
|
|
ΔGnps
|
-15.75 ± 0.83
|
|
ΔGbinding
|
-76.59 ± 10.23
|
To gain further insights into the major amino acid residue involved in binding, we have calculated the individual contribution of the residues across the entire length of the protofibril. It can be noted from Fig. 7a that chains A and B contributes the most towards the overall interaction energy. Subsequently, we have calculated the individual contribution of residues from these chains. As illustrated in Fig. 7b and c, our analysis reveals the key involvement of the non-polar hydrophobic residues - F19, A21, V24, and I32 of chains A and B.
The ability of BCL to interact with the hydrophobic residues of the core contributes to higher van der Waals interaction energy, as calculated by the MM-PBSA analysis. The animations (Online Resource 1 and 2) showcase the configurational changes in the protofibril structure throughout the 300 ns trajectory, both in Aβ-water and Aβ-BCL systems. The control system demonstrates slight twisting in the protofibril, whereas the presence of the BCL molecule significantly destabilises the protofibril structure. The contacts established by the BCL molecule with the protofibril residues at the 300 ns mark are illustrated in Supplementary Fig. S2. The aromatic ring of Phenylalanine and the side chain of Isoleucine residues play a vital role in ligand binding. Furthermore, the participation of the F19 residue in the protein-ligand interaction has been elucidated[76] in Supplementary Fig. S3. It was observed that initially, F19 formed π-stacking and hydrophobic interactions; however, towards the end, it predominantly exhibited hydrophobic interactions. This observation suggests that although other interactions play a role in the protein-ligand interaction, hydrophobic interactions ultimately drive the destabilisation process. The residue-wise energy contribution was also calculated for chains C, D and E, as depicted in Supplementary Fig. S7. Compared to chains A and B, these chains demonstrate a lower degree of interaction with the BCL molecule. The hydrophobic residues I32, L34, and V36 present in chains C, D and E contribute to their interaction with the BCL molecule.
The average BFE was also computed for the adjacent chains in both the presence and absence of the BCL molecule. The inter-chain BFE, detailed in Supplementary Table S3, indicated that in the presence of BCL molecule, the chains exhibited a higher BFE. This observation substantiates that, in the presence of the ligand, the adjacent chains of the protofibril displayed a diminished binding affinity, resulting in an increased distance between the chains.
We then carried out contact map analysis to qualitatively assess the effect of the ligand on the residue-residue contact between adjacent chains of the oligomer (Fig. 8). The contact map analysis provides the smallest distance between the Cα atoms of two residue pairs in Aβ42 protofibrils. The panel diagonals display the most substantial binding, representing homologous interactions between two neighbouring chains in close contacts, such as N- and C-termini from one chain with an equivalent portion from the neighbouring chain. The off-diagonals display the lateral non-homologous interaction, i.e. N-terminal interactions of one chain with the C-terminus of the other chain or vice-versa. The analysis was performed with the gmx mdmat module of GROMACS. The matrices were created using a minimum distance of 0–1.5 nm that separates the residue pairs. We have observed a drastic decrease in contact of the Aβ-BCL system compared to Aβ-water. The inter-chain contacts between Chain A, B and C were fairly compromised in the presence of the BCL molecule. The off-diagonals non-homologous interactions were mostly affected, as can be inferred from the colour range of the matrix shown in Fig. 8c and d. The contacts remained relatively sustained in Aβ-Water systems, as shown in Fig. 8a and b. The same trend in the inter-chain contacts was also observed between chains C, D and E, as depicted in Supplementary Fig. S8.
The compact oligomeric aggregates are produced by the strong inter-chain electrostatic and van der Waals interaction energies. The increased interchain distance, corroborated by the reduced inter-chain binding affinity, shows that the parent fibrillar structure in the Aβ-BCL systems moved from the highly ordered configuration towards a more destabilised form, with the chains further away. The MM-PBSA results and the decreased contact between the adjacent chains corroborate well, establishing BCL molecule as a promising destabilising agent of Aβ42.
Principal Component Analysis
Principal Component Analysis (PCA) serves as a multivariate statistical method employed to systematically reduce the necessary dimensions for describing protein dynamics. PCA operates as a linear transformation, extracting crucial elements in the data using a covariance matrix derived from atomic coordinates. PCA reads the MD simulation trajectory and extracts the dominant modes in the motion of the molecule. This reduction is achieved through a decomposition process that sifts observed motions from the most substantial to the smallest spatial scales. The analysis of principal components (PCs) can provide valuable insights into the predominant modes of motion exhibited by the protofibril in isolation and in complex with the BCL molecule. To illustrate alterations in motion induced in the protofibril by the interaction with BCL, PC analysis was conducted on the MD trajectories during the last 10 ns of simulation for Aβ42. Figure 9a exhibits plots of the eigenvalues against eigenvector indexes obtained from the diagonalization of the covariance matrix, where the eigenvalues reflect the intensity of the movements, and the eigenvector indicates movement directions. We observe that the amplitude of the first few eigenvalues decreased to reveal several constrained, more localized fluctuations. The analysis shows that the first, second, and third PCs account for 51.3%, 20.2%, and 9.35% in the Aβ-BCL system and 31%, 27.25%, and 12.7% in the Aβ-water system. The elevated eigenvalues observed in the Aβ-BCL system, in comparison to Aβ-water, suggest an increased magnitude of motion in the protofibril in the presence of the BCL molecule. To assess the impact of the BCL molecule on the motion represented by PC1, corresponding to the direction of maximum variance, the displacements of PC1 were calculated for both systems. As depicted in Fig. 9b, the results suggest that the BCL molecule influences the motions of the protofibril. In the control system, the motion is attributed to twisting, while in the presence of the BCL molecule, this motion is exacerbated. Notably, residues in Chain A and B regions exhibit elevated values, aligning with our MM-PBSA studies, where we observed that residues in these chains contribute significantly to the overall interaction.
Effect of BCL on various bonds of Aβ fibril
Intra- and inter-molecular bonds play a crucial role in maintaining the stability of protein structures. These bonds include hydrogen bonds, covalent bonds, salt bridges, and hydrophobic contacts. Understanding the changes in these molecular interactions in the presence of an external molecule would provide insights into its impact on protein stability. We have conducted a detailed analysis to elucidate the effect of BCL molecule on the critical molecular interactions that stabilise the Aβ42 (2BEG) structure.
Hydrogen Bond
Hydrogen bonds (H-bonds) are crucial in forming secondary structures and higher-order protein aggregates. They contribute significantly to structural integrity of protein. In Aβ fibrils, there is an extensive intra- and inter-peptide H-bonding network. Several experimental and theoretical investigations have demonstrated the importance of these H-bonds in stabilising Aβ fibrils [69, 77].
We have calculated the average number of H-bond over 300 ns trajectory for the entire protein. The number of H-bond has been reduced from 59 ± 5 to 52 ± 5. The decreasing trend of the average number of H-bond can also be observed in Fig. 10. The average inter-chain H bond has also been tabulated in Supplementary Table S4. There is an observable decrease in H bonds between chains A-B and B-C in Aβ-BCL systems. The number decreased from 11 ± 2 to 9 ± 1 between chains A-B and from 15 ± 1 to 10 ± 1 between chains B-C. The reduction in H-bond indicates the increased instability of the protofibril. These observations are in close agreement with the destabilisation of Aβ fibril by a proline-rich β-sheet breaker peptide [78].
Salt Bridge
A salt bridge is formed between two oppositely charged amino acid residues, typically between an acidic residue such as glutamate or aspartate and a basic residue such as arginine, lysine or histidine. The interaction between these residues results in the formation of an electrostatic bond. Salt bridges are essential structural features of proteins, and play a critical role in stabilising the three-dimensional structures of the protein [79]. In the protofibril structure chosen for the current study, the salt bridges formed between D23 and K28 residues are more prominent than those formed between E22 and K28 residues[37].We report only those significant interchain salt bridges (viz., D23 and K28), by measuring the average distance between D23 and K28.
It can be observed from Fig. 11 that the average D23-K28 distances for all the four sets of neighbouring chains increased across the trajectory for the Aβ-BCL system. In contrast, the distance for the Aβ-Water system does not show much changes, rather remains relatively stable. Although, there was an increase in the average D23-K28 distance for chain A even in the control system, it was less prominent than in the Aβ-BCL system. The increase in average distance for chain A in the control system was because of the inherent configuration of the K28 residue. The K28 residue of chain A points away from the D23, resulting in a weaker electrostatic bond. This weaker electrostatic bond explains the slight outward movement of chain A, as has been observed in Fig. 3a. However, the Aβ-BCL system shows a notable increase in the average distance between the D23 and K28 residues of all other adjacent chain pairs compared to the control systems. The increased distance signifies the weakening and disruption of these inter-chain salt bridges, which are crucial for the overall stability of the protofibril.
Hydrophobic contacts
Besides electrostatic interactions, hydrophobic interactions also play a crucial role in protein folding and stability. The hydrophobic residues buried inside protein cores enhance protein stability. Studies on Aβ fibrils have reported the vital role of hydrophobic contacts in the structural organisation and strengthening of the fibrils [37]. The hydrophobic contact pairs A21-V36, L34-V36, and F19-G38 have been identified as significant contributors towards the stability of the current Aβ pentamer structure [34].
As discussed earlier, the MM-PBSA analysis revealed that the BCL molecule firmly binds to the F19 and A21 residues of chain A and B. We assumed that the binding of the BCL molecule to F19 and A21 residues may have disrupted the crucial hydrophobic bonds associated with these residues. Therefore, we calculate the average distances between the inter-chain A21-V36 and F19-G38 residues to study the disruption of hydrophobic contacts, as the interchain residue distance measures the compactness of the protofibril structure.
As observed from Fig. 12, all the average inter-chain A21-V36 and F19-G38 distances has increased significantly in the presence of the BCL molecule. The only deviation was observed in chain A, where the distance between the A21-V36 residues was slightly higher in the Aβ-Water system. As explained earlier, the orientation of the K28 residue of chain A in the native protofibril model results in a weaker electrostatic bond between chains A and B, making chain A more susceptible to fluctuations. Therefore, this weaker electrostatic bond between chains A and B, explains the observed increase in the average A21-V36 distance, even for the Aβ-Water system. Hence, by binding to the hydrophobic residues, the BCL molecule has successfully destabilised the protofibril. In-silico study of the wgx-50 molecule has yielded similar results, demonstrating that disrupting these crucial hydrophobic bonds can destabilise the Aβ fibrils[36].
{kind=link}