Exosomes derived from M1 macrophages are mainly phagocytosed by microglia in vivo and vitro
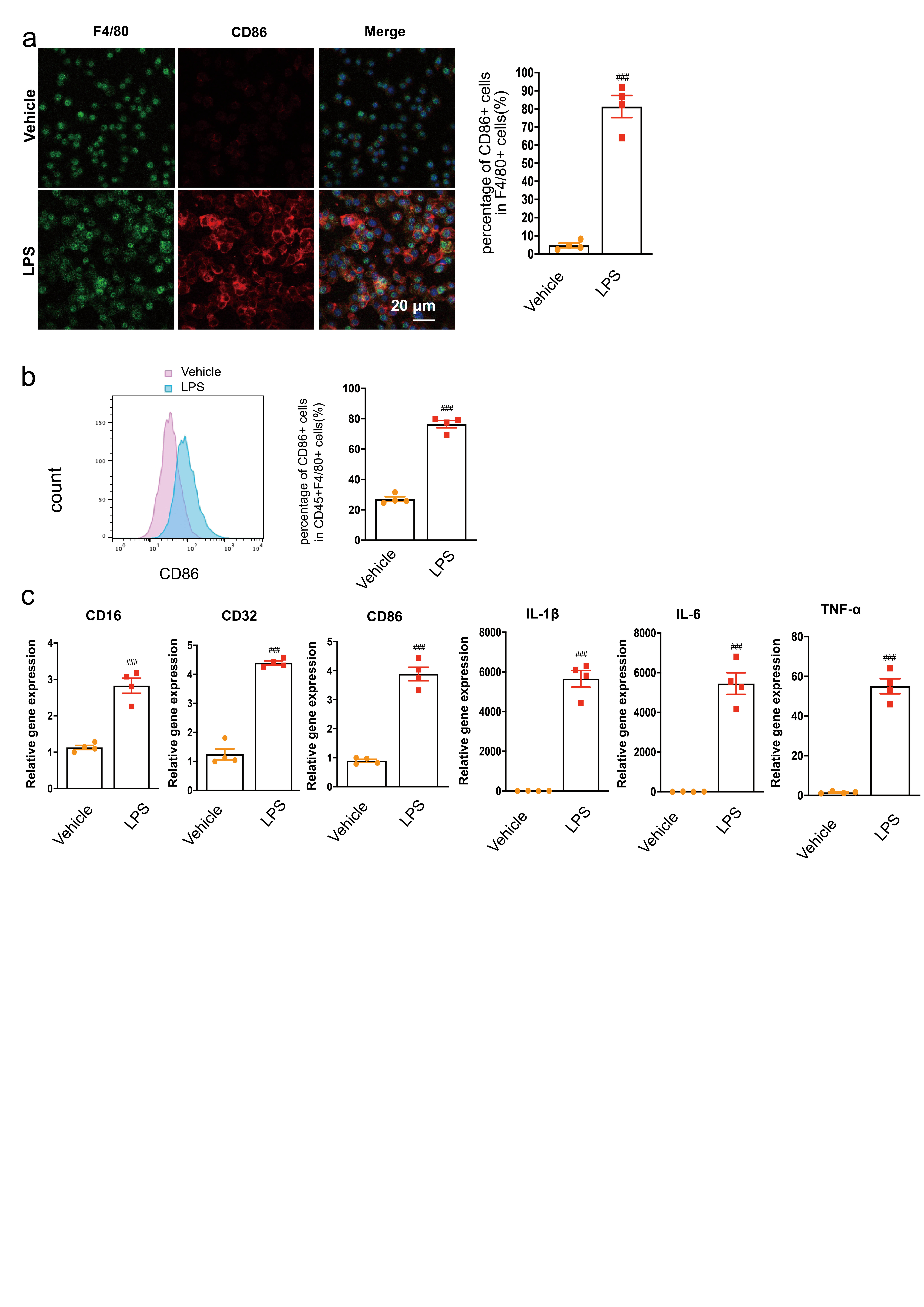
To explore the role of M1 macrophage-derived exosomes, we employed LPS treatment on RAW264.7 cells to induce M1 macrophages. Immunofluorescence, RT-PCR, and flow cytometry analyses revealed that LPS effectively induced the polarization of M1 macrophages. (Fig. S1a-c). Exosomes were isolated from the supernatants of M1 macrophages using ultracentrifugation. Nanoparticle tracking analysis (NTA) revealed that the diameter of the exosomes predominantly ranged from 60 to 160 nm (with a mean of 141.8 ± 3.4 nm), as depicted in Fig. 1a. Electron microscopy images illustrated the characteristic cup-shaped morphology of the exosomes (Fig. 1a). Western blot analysis confirmed the presence of exosomal markers, including CD63 and TSG101, in the exosomes derived from M1 macrophages (Fig. 1b).
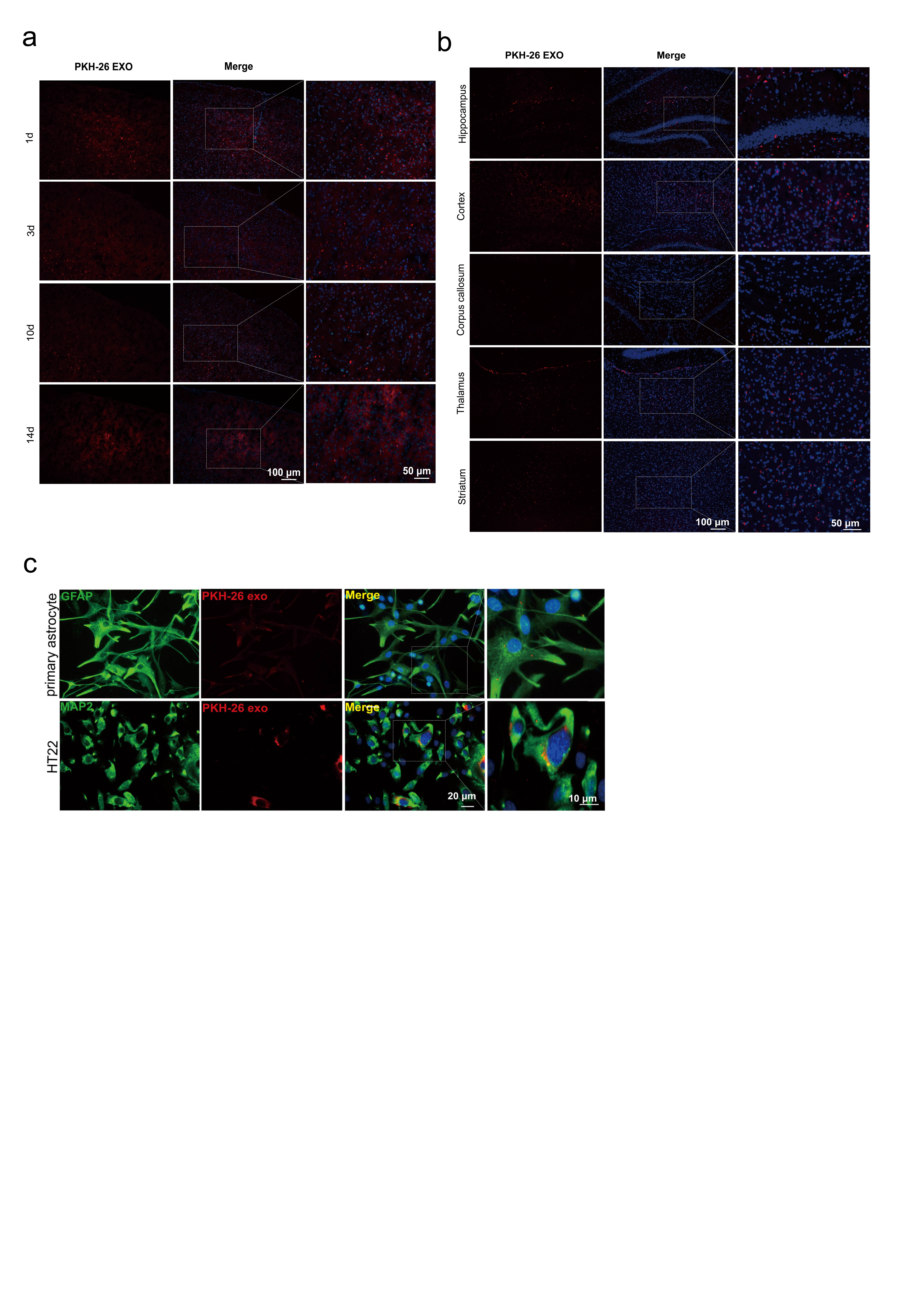
The exosomes derived from M1 macrophages, termed M1-EXO, were labeled with PKH26 (red) to investigate their ability to cross the blood-brain barrier and their distribution and metabolism in the brain of APP/PS1 mice following tail vein injection of 100 µg. Remarkably, PKH26-labeled red M1-EXO were detected in the brain as early as one day post-injection, persisting up to 14 days later (Fig. S2a). These exosomes exhibited predominant distribution within the cortex, hippocampus, and striatum, with a lesser presence in the thalamus, and minimal distribution observed in the corpus callosum (Fig. S2b).
Next, we further investigated whether M1-EXO were internalized by microglia, astrocytes, oligodendrocytes, and neurons in the cortex and hippocampus. We observed that M1-EXO (marked in red) were predominantly internalized by iba1 + microglia (highlighted in green), with a smaller portion being taken up by GFAP + astrocytes and MAP + neurons, while minimal uptake was observed by Olig2 + oligodendrocytes in the cortex and hippocampus (Fig. 1c,d). To further explore the phagocytic behavior of M1-EXO, we conducted experiments using BV2 cells and primary microglia in vitro. After 24 hours of treatment, both BV2 cells and primary microglia, labeled positively for Iba1 (shown in green), demonstrated internalization of M1-EXO (marked in red) (Fig. 1e). These in vitro findings are consistent with our in vivo observations, demonstrating the effective uptake of M1-EXO by microglia. Additionally, our study revealed that astrocytes and neurons internalized only a small fraction of exosomes in vitro(Fig. S2c).
M1-EXO exacerbated cognitive impairment and facilitated the deposition of Aβ in APP/PS1 mice.
To investigate the impact of peripheral exosomes on cognitive function and Aβ deposition in APP/PS1 mice, two types of exosomes (M0-EXO and M1-EXO) were administered via tail vein injection every 2 weeks for a total of 4 injections. Two weeks after the final exosome injection, Morris water maze (MWM) and open-field tests were conducted. In the MWM test (Fig. 2a), M1-EXO significantly impaired spatial learning and memory in APP/PS1 mice, evidenced by increased latency to reach the platform from days 1 to 5, and reduced platform frequency during the probe trial on day 6 (Fig. 2b). Additionally, we employed the open-field test to evaluate the effects of M1-EXO on exploration-related behaviors. Compared to APP/PS1 mice receiving M0-EXO, those treated with M1-EXO exhibited notable deficits in exploration ability, spending less time in the center and making fewer central entries (Fig. 2c,d). Furthermore, the total distance moved and average speed of movement in APP/PS1 mice receiving M1-EXO were inferior to those in mice receiving M0-EXO (Fig. 2d).
Aβ deposition and neuronal loss are pivotal pathological hallmarks of AD. Our study aimed to investigate whether M1-EXO accelerates Aβ deposition and induces neuronal death. Comparing APP/PS1 mice treated with M0-EXO to those treated with M1-EXO, we observed a significant increase in Aβ plaque(Thios+) density in both the cortex (by 18.6%) and hippocampus (by 19.8%) in the M1-EXO-treated group (Fig. 2e,f). Additionally, M1-EXO treatment was associated with elevated microglial proliferation (Fig. 2e, g). Immunofluorescence revealed a reduction in NeuN-positive neurons in the cerebral cortex and hippocampus following M1-EXO administration (Fig. 2h). M1-EXO exacerbated cognitive impairment, facilitated Aβ deposition, accelerated neuronal death, and induced microglial proliferation in APP/PS1 mice.
M1-EXO induced the accumulation of disease inflammatory microglia (DIM) in APP/PS1 mice
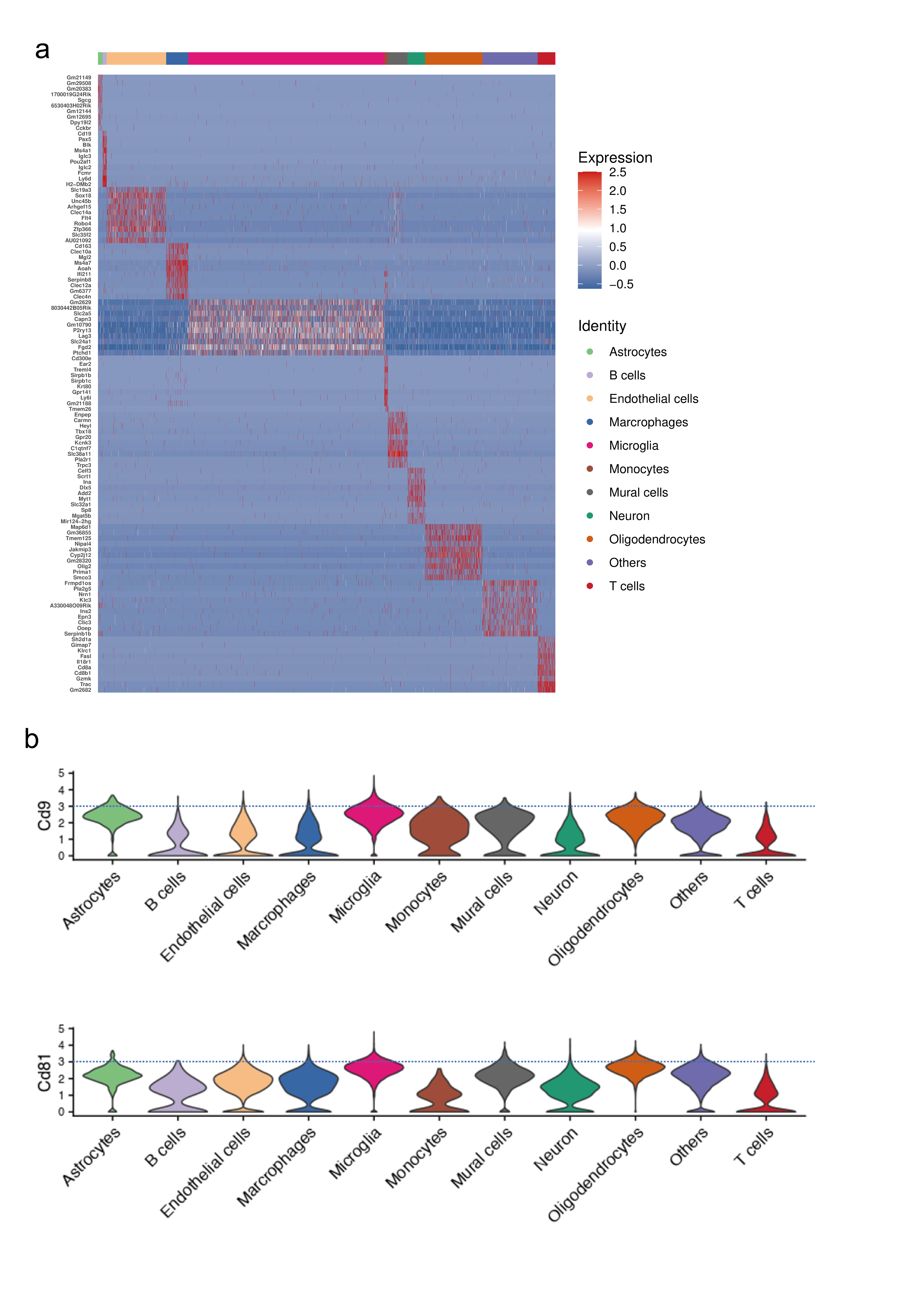
To comprehensively characterize changes in the major cell types within the brains of APP/PS1 mice following M1-EXO administration, we utilized droplet-based single-cell RNA sequencing (10× Genomics) on cells isolated from the cerebral cortex and hippocampus of two lines of APP/PS1 mice treated with M1-EXO, along with two lines treated with M0-EXO. Subsequently, we performed unsupervised clustering and annotated cell types based on canonical gene markers. As illustrated in the UMAP plot, our analysis identified 11 distinct cell types, including astrocytes, B cells, endothelial cells, macrophages, microglia, monocytes, mural cells, neurons, oligodendrocytes, T cells, and others(Fig. 3a). We identified marker genes for each cell-type and displayed the top 10 marker genes in a heatmap (Fig. S3a). Notably, microglia constituted the majority of cells, and we observed a significant increase in microglia following M1-EXO administration in APP/PS1 mice (Fig. 3b). The proliferation of microglial cells may be linked to cognitive impairment and Aβ deposition in the M1-EXO group.
Given the critical role of microglia in brain function and their involvement in exosomal phagocytosis, our study focused on microglia at the single-cell level. Single-cell sequencing results demonstrated high expression of Cd9 and Cd81 (exosomal markers) in microglia, consistent with the predominant internalization of EXO by microglia(Fig. S3b). To further explore this, we performed differential gene expression analysis to identify specific marker genes for subclusters within microglia. We identified three distinct subclusters for further investigation(Fig. 3c). According to the subcluster proportional diagram, we observed a significant increase in the proportion of cluster 2 (12.2%) following M1-EXO administration. In contrast, there was a decrease in the proportion of cluster 3 (10%), while cluster 1 showed minimal change (Fig. 3d).
Gene expression analysis was conducted to identify marker genes specific to individual subclusters. We generated a heatmap displaying the top marker genes for each subcluster (Fig. 3e). This analysis revealed that a substantial number of highly expressed genes demonstrated distinct patterns within their respective microglial subclusters, underscoring unique expression profiles. Cluster 1 exhibited high expression of genes associated with Disease-Associated Microglia (DAM), including Spp1, Lpl, Cst7, Ccl6, Lyz2, Ctsd, Axl, Trem2, and Apoe (Fig. 3e and f). DAM are recognized as a distinct microglial subtype linked to AD. Cluster 2 demonstrated significant expression of inflammatory marker genes previously reported in the literature (Hspa1a, Ccl2, Ccl3, Ccl4, Ccl12, Cxcl10), as illustrated in Fig. 3e and g. Cluster 3 showed elevated expression of microglial homeostatic genes (Hao1, Tmx4, Tmem119, Crybb1, Hpgd, Glul, P2ry12), as depicted in Fig. 3e and h. This indicates a predominant homeostatic state among microglia within this cluster. These findings underscore the heterogeneity of microglial subtypes and their differential responses, providing insights into their functional roles in the context of M1-EXO administration.
In a previous study, comparative analysis of gene expression data from Hammond, Keren-Shaul, and Van Hove datasets identified a conserved signature termed DIM[26], encompassing genes associated with pro-inflammatory responses and immune activation (Table S2). Cluster 2 cells exhibited a broad range of DIM scores calculated using AddModuleScore, indicating an elevated inflammatory state within this cell population (Fig. 4a). Based on the heightened expression levels of inflammation-related genes and their prevalence in neurodegenerative conditions, we designated this subset as disease inflammatory microglia (DIM). Expression analysis showed upregulation of several representative genes (Il1b, Il1a, Cd86, Tnf, Ccl4, Btg2, Ier2, Junb, Ccl12, Cd14) in DIM, contrasting with their downregulation in homeostatic microglia, thus highlighting the distinct inflammatory signatures of DIM (Fig. 4b and c).
Based on extensive in vivo studies on M1/2 microglia, we observed that "M1" microglial markers such as Cd86 and Cd14 were predominantly expressed in DIM (Fig. 4b). In contrast, "M2" markers like Arg1 and CD206 (encoded by Mrc1) were absent in microglia. However, our scRNA-seq data revealed that Cd86 was expressed in both microglia and macrophages, whereas Mrc1 was predominantly expressed in macrophages (Fig. 4e). This suggests that infiltrating macrophages may have been misclassified as traditionally defined "M2" microglia. Therefore, the conventional M1/M2 microglia categorization might not effectively distinguish between distinct microglial subpopulations or accurately depict their roles in APP/PS1 mice.
We further validated the impact of M1-EXO on DIM formation through in vivo experiments. Immunofluorescence analysis revealed a notable increase in microglia and the proportion of CD86-positive microglial cells in the cortex and hippocampus of brain tissue from APP/PS1 mice, suggesting that M1-EXO induced the formation of DIM cells (Fig. 5a, b). Additionally, M1-EXO stimulated the RNA expression of disease-associated inflammatory markers CD16, CD32, CD86, TNF-α, IL-6, and IL-1β in these brain regions (Fig. 4a, b). These findings underscore the role of M1-EXO in promoting the emergence of DIM and enhancing the expression of inflammatory markers associated with AD.
M1-EXO induce the transformation of homeostatic microglia into DIM and triggered phosphorylation of the JAK1/STAT1 signaling pathway.
Following the injection of M1-EXO, we observed a decrease in the proportion of homeostatic microglia and a concurrent increase in DIM, while the proportion of DAM did not change significantly. To explore the dynamic immune states and cell transitions, we conducted pseudotime analysis, excluding DAM cells. This analysis revealed that homeostatic microglia were situated at the beginning of the pseudotime trajectory, while DIM cells occupied a terminal state (Fig. 6a). We further explored the transcriptional changes associated with transitional states and noted that this process could be categorized into 3 distinct phases (Fig. 6b). A gene expression heatmap across pseudotime showed an upregulation of the DIM-conserved signature (including Ccl12, Ccl5, Cd14, Cxcl10, Il1a, Il1b, Nfkbia, Tnf), whereas the homeostatic signature, despite an initial increase, was downregulated during these transitions (Fig. 6b, c). Therefore, the results suggest that M1-EXO induces the transformation of homeostatic microglia into DIM. Pathway analysis indicated enrichment of inflammation-related signaling pathways in phase 3 (Fig. 6b). For functional validation, we performed gene set variation analysis (GSVA) in the APP/PS1 + M0-EXO and APP/PS1 + M1-EXO groups. Notably, ‘JAK-STAT signaling pathway’ was at the forefront of our findings, providing a crucial clue about the mechanisms by which M1-EXO induces DIM formation. The JAK-STAT pathway is crucial in cellular communication, modulating inflammatory responses triggered by cytokines, growth factors, and inflammatory signals like LPS[27]. JAK1, which can be phosphorylated by interleukin-6 (IL-6), IFN-γ, and other factors, is widely expressed across various tissues and has the ability to phosphorylate all STAT proteins[28]. Specifically, STAT1 activation can be triggered by interferons (IFN), IL-2, IL-6, and tumor necrosis factor (TNF)[29]. Previous studies have indicated that activated STAT1 initiates the transcription of M1-associated genes, leading to the production of inflammatory cytokines[30, 31]. Given the pivotal role of JAK1/STAT1 in inflammatory responses and immune regulation, we have chosen to focus on it as the primary subject of our study.
Based on insights from single-cell sequencing, we conducted Western blot validation to assess the activation status of JAK1/STAT1 in the cortex and hippocampal regions. Our results demonstrated that M1-EXO induced phosphorylation of the JAK1/STAT1 signaling pathway without altering the overall expression levels of JAK1/STAT1 (Fig. 6e).
To elucidate the role of M1-EXO, we employed primary microglia for in vitro experiments. Microglia were co-cultured with exosomes for 24 hours to examine the biological effects of M1-EXO on DIM formation. As depicted in Fig. 7a, there was a notable increase in the proportion of DIM(CD86+) in the M1-EXO group compared to the M0-EXO group. Furthermore, flow cytometry analysis of CD86-positive cells revealed that M1-EXO significantly induced microglial polarization towards DIM(Fig. 7b). Additionally, the RNA expression levels of CD16, CD32, CD86, TNF-α, IL-6, and IL-1β (associated with DIM phenotype) were significantly elevated (Fig. 7c). These results collectively indicate that M1-EXO effectively triggers the formation of DIM. p- We further analyzed the activation status of the JAK1/STAT1 signaling pathway. We found notable upregulation of p-JAK1 and p-STAT1 in the M1-EXO group, whereas there was no significant difference in the expression levels of JAK1 and STAT1 in M1-EXO-treated microglia compared to controls (Fig. 7d). Our study, utilizing both in vivo and in vitro experiments, confirms that M1-EXO induces the transformation of homeostatic microglia into DIM and activates the phosphorylation of JAK1/STAT1 signaling pathway.
miR-155-5p is upregulated in M1-EXO and can be transferred to microglia by exosomes.
To explore the mechanisms underlying DIM formation induced by M1-EXO, we conducted miRNA sequencing of exosomes, as miRNAs are a major component of exosomes. Compared to M0-EXO, 4 miRNAs (miR-155-5p, miR-146b-5p, miR-21a-5p, miR-1843b-5p) were upregulated, while 12 miRNAs (miR-204-3p, miR-23a-5p, miR-5099, etc.) were downregulated in M1-EXO (Fig. 8a, b). Subsequent validation by RT-PCR revealed that miR-155-5p was the significantly upregulated microRNA in M1-EXO (Fig. 8c). Based on these findings, we decided to investigate the potential roles of miR-155-5p in the neuroinflammatory response of microglial cells. We co-cultured exosomes with primary microglial cells and monitored changes in miR-155-5p expression. Following a 24-hour treatment of primary microglial cells with exosomes, we observed an approximately 4.5-fold increase in miR-155-5p levels in M1-EXO group (Fig. 8d).
This finding suggests that M1-EXO may exert its effects through the carried miR-155-5p. Therefore, we utilized lentiviral vectors to knock down miR-155-5p (miR-155KD) in RAW264.7 cells. As expected, a significant reduction in miR-155-5p expression was observed in miR-155KD RAW264.7 cells compared to miR-NCKD RAW264.7 cells (Fig. 8e). Following LPS stimulation, we isolated exosomes released from both miR-155KD and miR-NCKD RAW264.7 cells. Subsequent analysis confirmed a substantial decrease in miR-155-5p levels within the exosomes derived from miR-155KD RAW264.7 cells (Fig. 8f). Therefore, we extracted M1 miR-NCKD EXO and M1 miR-155KD EXO exosomes for further validation experiments.
M1-EXO induced DIM formation by delivering miR-155-5p in vivo and vitro.
We explored the effects of exosome-carried miRNA-155-5p on cognition, Aβ deposition, DIM formation, and associated pathways. Initially, miR-NCKD RAW264.7 and miR-155KD RAW264.7 cells were separately stimulated with LPS to produce M1 miR-NCKD EXO and M1 miR-155KD EXO. To assess whether miR-155-5p modulation in M1-EXO affect behavioral deficits in APP/PS1 mice, Morris Water Maze (MWM) tests were conducted 2 months after exosome administration. Our findings demonstrated that treatment with M1 miR-155KD EXO enhanced spatial learning and memory compared to administration of M1 miR-NCKD EXO (Fig. 9a). Additionally, we observed that knocking down miR-155-5p in M1 EXO reduced Aβ deposition (Thios+) in the cortical and hippocampal regions (Fig. 9b, c). M1 miR-155KD EXO administration resulted in decreased DIM formation and inhibited mRNA expression of CD16, CD32, CD86, IL-1β, IL-6, and TNF-α compared to M1 miR-NCKD EXO administration (Fig. 9d,e,f). Moreover, administration of M1 miR-155KD EXOs reduced the phosphorylation of JAK1/STAT1 in APP/PS1 mice compared to M1 miR-NCKD EXO administration (Fig. 9g).
Cellular experiments further validated the role of miRNA-155-5p in M1-EXO-induced DIM formation. We co-cultured primary microglial cells with M0 EXO, M1 miR-NCKD EXO, and M1 miR-155KD EXO for 24 hours. Immunofluorescence staining experiments revealed that M1 miR-155KD EXO reduced the percentage of CD86 + cells among iba1 + cells compared to M1 miR-NCKD EXO (Fig. 10a). Flow cytometry analysis of CD86 expression in primary microglia treated with these exosomes confirmed these findings (Fig. 10b). RT-PCR results also indicated that M1 miR-155KD EXO reduced the mRNA expression of CD16, CD32, CD86, IL-1β, IL-6, and TNF-α compared to the M1 miR-NCKD EXO (Fig. 10c). Additionally, western blot analysis validated that M1 miR-155KD EXO reduced the phosphorylation of JAK1/STAT1 compared to M1 miR-NCKD EXO in primary microglia (Fig. 10d). Taken together, our results suggest that M1 macrophage-derived exosomes deliver miR-155-5p to promote DIM formation by modulating the phosphorylation of JAK1/STAT1 signaling pathways.
M1 exosomal miR-155-5p induces DIM formation by downregulating SOCS1 protein expression and activating the phosphorylation of the JAK1/STAT1 signaling pathway
We further explored the mechanisms by which miR-155-5p, derived from exosomes released by M1 macrophages, induces DIM expression. Data from PicTar, miRanda, TargetScan, and the miRDB database revealed an alignment between the miR-155-5p sequence and the 3’-UTR sequence of SOCS1, indicating that miR-155-5p may target SOCS1 to induce DIM formation(Fig. 11a). Previous studies have reported that SOCS1, a suppressor of cytokine signaling (SOCS1), is targeted by miR-155-5p, regulating inflammation and immunity via the JAK-STAT pathway[32, 33]. To further confirm that the SOCS1 3’-UTR is a direct target of miR-155-5p, we conducted luciferase experiments. Our study showed that miR-155-5p overexpression significantly reduced luciferase activity when co-transfected with the WT-3’ UTR of SOCS1 compared to the control(Fig. 11b). In contrast, no inhibitory effect of miR-155-5p on luciferase activity was observed when co-transfected with the MUT-3’ UTR of SOCS1(Fig. 11b). We also observed higher expression of SOCS1 protein in the cortex and hippocampus of APP/PS1 mice treated with M1 miR-155KD EXO compared to those treated with M1 miR-NCKD EXO (Fig. 11c). These findings suggest that miRNA-155-5p carried by M1-EXO may induce DIM formation and initiate inflammatory responses by regulating SOCS1 expression. Next, to investigate the role of SOCS1 in M1-EXO induced DIM formation, we employed an AAV mediated approach targeted specifically at microglia to overexpress SOCS1 in the brains of APP/PS1 mice. Following a 1-month injection of AAV(red), immunofluorescence experiments confirmed successful infection of microglia(green) in the cerebral cortex and hippocampus of APP/PS1 mice (Fig. 11d and e). Further analysis through western blot showed that in the APP/PS1 + AAV-SOCS1 group, SOCS1 protein levels in both the cerebral cortex and hippocampus were notably elevated by 43.3% and 42.7%, respectively, in comparison to the APP/PS1 + AAV-CON group.
Subsequent experiments validated that SOCS1 overexpression significantly alleviated cognitive deficits and Aβ deposition induced by M1-EXO administration in APP/PS1 mice (Fig. 12a, b and c). To investigate whether SOCS1 overexpression ameliorates cognitive deficits by suppressing the phosphorylation of the JAK1/STAT1 signaling pathway, our findings showed a significant increase in the phosphorylation of JAK1 and STAT1 in the cerebral cortex and hippocampus of M1-EXO-administered APP/PS1 + AAV-CON mice, indicating activation of this signaling pathway. However, SOCS1 overexpression effectively attenuated the phosphorylation of JAK1 and STAT1. These results collectively suggest that SOCS1 overexpression can mitigate cognitive deficits and Aβ deposition by suppressing the phosphorylation of the JAK1/STAT1 signaling pathway in M1-EXO-administered APP/PS1 mice.
{kind=link}
{kind=link}
{kind=link}