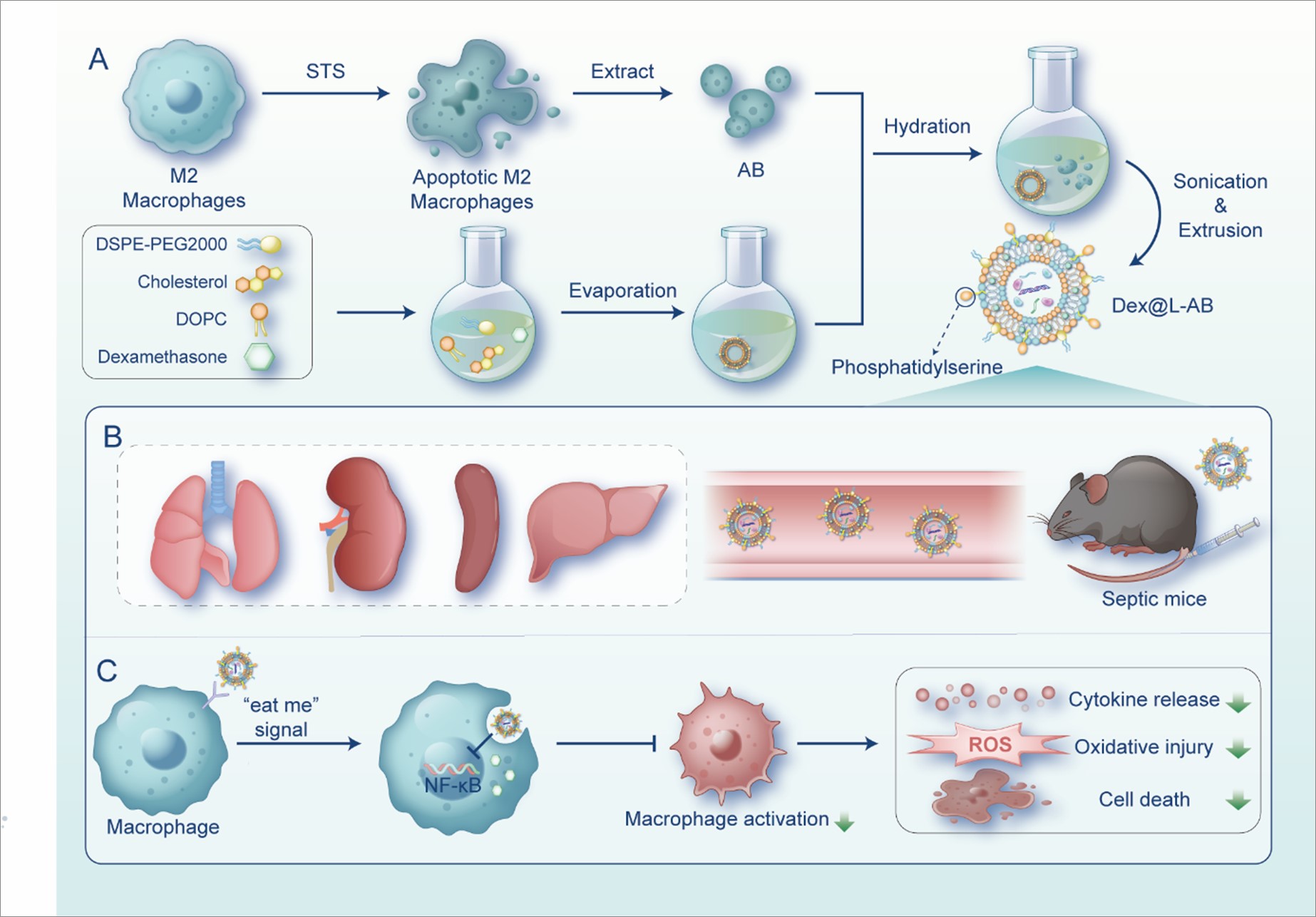
Preparation and characterization of L-AB
The AB were combined with liposomes to fabricate L-AB. A lipid film was prepared using DOPC, cholesterol, and DSPE-PEG2000 at a molar ratio of 8:8:1 by the solvent evaporation method. Subsequently, the AB suspension was hydrated with the prepared lipid film and further extruded through a 1 µm polycarbonate membrane to produce L-AB. The morphological features of AB, LIP and L-AB were observed by transmission electronic microscopy (TEM). As displayed in Fig. 1A, the AB exhibited a larger spherical vesicle structure with a particle size of approximately 1000 nm, in contrast to LIP and L-AB, which had significantly smaller particle sizes of approximately 200 nm and also showed a similar spherical vesicle structure. Subsequently, the particle size, polydispersity index (PDI), and ζ potential of the nanovesicles were determined by dynamic light scattering (DLS), and the results were shown in Fig. 1B. Consistent with TEM results, the size of L-AB was around 151.07 ± 2.96 nm (PDI, 0.222 ± 0.025), while those of LIP and AB were 169.27 ± 2.48 nm (PDI, 0.215 ± 0.023) and 984.84 ± 13.87 nm (PDI, 0.383 ± 0.047), respectively. Besides, the decreased PDI indicated that L-AB possessed a better size homogeneity than AB. There was a decreased ζ potential of L-AB (-5.05 ± 2.20 mV) compared to LIP (-0.15 ± 3.08mV), probably due to the insertion of AB membrane proteins (-9.95 ± 2.70 mV). The colloidal stability of LIP, AB and L-AB was further evaluated by monitoring the particle size over a period of 7 days. Both LIP and L-AB demonstrated stability over a period of one week in biologically relevant solutions, as indicated by minimal changes in particle size. In contrast, AB exhibited a notable change in particle size, with a reduction from 984 to 531 nm over the subsequent day (Fig. S1). The results indicated that the fusion of AB with the synthetic liposomes favored the structural stability of AB. The findings indicated that fusion of AB with synthetic liposomes resulted in improved structural stability of AB.
Förster resonance energy transfer (FRET) was employed to confirm the fusion of LIP and AB, utilizing DiI (a fluorescent receptor) and DiD (a fluorescent donor). DiI and DiD co-labeled AB were fused with blank LIP in different ratios. As shown in Fig. 1C, an increase in the quantity of LIP was accompanied by an enhancement in fluorescence at 565 nm, while a decline was observed at 670 nm. These results indicated that the LIP and AB can be fused into nanovesicles. Similarly, LIP and AB were respectively labeled with red (DiI) and green (DiO) fluorescent dyes and imaged using a confocal laser scanning microscope (CLSM) to validate co-fusion efficiency. As shown in Fig. 1D, physically mixed DiI@LIP and DiO@AB exhibited a segregated distribution. Following the execution of hydration and coextrusion, the red (DiI) and green (DiO) fluorescence in L-AB was largely overlapped. In the merged fluorescence, DiI@LIP and DiO@AB showed colocalization, indicating that the liposomes and AB had fused.
Fourier transform infrared (FTIR) spectroscopy confirmed the hybridization of LIP, AB, and L-AB by analyzing their vibration fingerprints. As shown in Fig. 1E, the infrared spectra of lipids, carbohydrates and proteins showed overlapping results within the 4000 − 400 cm− 1 range, with symmetrical stretching vibrations of C = C and C = O at 1700 − 1600 cm− 1. The AB sample exhibited a single peak of C = O at 1650 cm− 1. L-AB showed enhanced absorption at 1650 cm− 1 compared to LIP, indicating integration with AB. Overlapping with LIP in the 1500 − 1350 cm− 1 range suggested absorption of lipid hydrocarbons. Protein-related sugar chains were observed in the 1200 − 900 cm− 1 region, with L-AB exhibiting a peak shape similar to that of AB, which indicated the presence of glycosylated proteins. Thus, the FTIR results illustrated that L-AB inherited functional groups of protein compositions from AB.
Furthermore, the surface of AB exhibited a notable presence of PS, a molecule known for its strong binding affinity to Annexin V, as illustrated in Fig. S2. To assess the prevalence of PS and CD11b (a macrophage surface protein) on the surface of L-AB, nanoflow cytometry was employed in the investigation. The results revealed a high expression of PS (63.4%) and CD11b (46.0%) on L-ABs, as depicted in Fig. 1F. The results demonstrated that the L-AB acquired biological components from M2 macrophages as a consequence of their collaboration with AB, which may facilitate the execution of anti-inflammatory functions.
In further study, AB and L-AB were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) to examine the protein preservation. As shown in Fig. 1G, the protein profiles of the M2 macrophages were similar to those of the original M0 cells with minor variations. The protein profiles of AB and L-AB were analogous to the total protein band of M2 cells that they were derived from, showing that proteins had been retained fairly intact. Specifically, the expression of CD206 was characterized through Western blot, which is the respective marker of M2 macrophages. The CD206 expression level in M2 cells was found to be increased in comparison to that observed in M0 cells. AB exhibited the presence of CD206 proteins derived from M2 macrophages, and the EV marker CD9 was expressed at an elevated level in AB. Furthermore, high levels of apoptosis-associated markers, cleaved caspase-3, were observed (Fig. 1H). Concurrently, L-AB expresses major proteins at comparable levels to AB, which may exhibit biological functions analogous to those of AB.
Cellular uptake and anti-inflammatory ability at the cellular level
Firstly, we examined the cell viability of LIP, AB, and L-AB on RAW264.7 through 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. As indicated in Fig. S3, no obvious cytotoxic effects were observed and L-AB displayed safety on RAW264.7 cells. Subsequently, the uptake characteristics of AB, LIP and L-AB by macrophages were evaluated. A significant red fluorescence signal was observed in the AB and L-AB groups compared to that in the LIP groups (Fig. 2A), indicating that AB and L-AB were taken up by macrophages with high efficiency. This phenomenon may be attributed to the clearance of AB by macrophages. Next, the time-dependent uptake of L-AB by macrophages was examined. As anticipated, the fluorescence intensity exhibited a notable increase with the duration of the incubation period. The mean fluorescence intensity (MFI) of the L-AB group was observed to be greater than that of the LIP group at 1 h, although this difference was not statistically significant. In contrast, the MFI of the L-AB group was significantly higher than that of the LIP group at 3 and 5 h. This disparity may be attributed to the presence of PEG on the surface of the L-AB, which potentially hindered macrophage uptake and consequently prolonged its circulation time in vivo Fig. 2B-C. Moreover, the enhancement in the uptake of AB and L-AB by macrophages was more pronounced. The MFI of macrophages ingesting AB was significantly higher than that of LIP and L-AB. This may be attributed to the size of AB, which was significantly larger than that of LIP and L-AB, and more fluorescent dyes were ingested by macrophages phagocytizing larger volumes of nanoparticles under the same conditions. To confirm the targeted uptake of L-AB by macrophages, control recipient cells were selected from AML12 (alpha mouse liver 12), HUEVC (human umbilical vein endothelial cells), L929 (mouse fibroblast), and MLE12 (mouse lung epithelial cells). The findings indicated that macrophages treated with DiD-labeled L-AB exhibited significantly greater uptake compared to AML12, HEUVC, L929, and MLE12 when subjected to identical conditions (Fig. 2D). This result indicates that L-AB can be specifically taken up by macrophages. It is therefore postulated that L-AB are primarily internalized by macrophages within bodily tissues.
Dex, a hydrophobic anti-inflammatory agent, was employed as a model molecule for evaluating the efficacy of drug delivery by L-AB. The analysis of the encapsulation efficiency of Dex within L-AB was conducted using high-performance liquid chromatography (HPLC). The results demonstrated an encapsulation efficiency of 68.66 ± 7.74% for Dex within L-AB (Fig. S4). Furthermore, the encapsulated Dex exhibited a sustained release pattern, with approximately 78.5% of the drug released from the L-AB over a 48h period (Fig. 3A).
A cytokine storm frequently results in an excessive production of reactive oxygen species (ROS), which can lead to oxidative damage and the amplification of inflammation [33, 34]. The impact of Dex@L-AB on the protection against intracellular endogenous ROS (induced by Lipopolysaccharides (LPS) stimulation) was examined. As shown in Fig. 3B-C, the intracellular ROS level, as quantified by 2′,7′-dichlorofluorescein diacetate (DCFH-DA), exhibited a marked increase following LPS treatment. In contrast, the MFI of DCFH-DA was significantly reduced in cells treated with Dex, indicating an enhanced ROS scavenging activity of Dex. Furthermore, the ROS level in the Dex@L-AB group was unexpectedly slightly lower than that in the Dex group. This may be due to the activation of the anti-inflammatory pathway of macrophages by the stimulation of AB. Besides, the levels of NO were found to be decreased following the administration of AB, Dex, Dex@LIP or Dex@L-AB, which indicated that both AB and Dex were capable of downregulating NO production (Fig. 3D).
Next, the impact of Dex@L-AB on the inhibition of pro-inflammatory cytokines was further investigated by measuring the levels of IL-6, interferon-γ (IFN-γ), and TNF-α (Fig. 3E-G). The cytometric bead array (CBA) analysis demonstrated a significant increase in cytokine secretion by LPS-stimulated macrophages, and drugs such as Dex and AB were able to inhibit these secretions to varying degrees. As anticipated, the Dex@L-AB group exhibited the most pronounced inhibitory effect on pro-inflammatory factors.
The nuclear translocation of nuclear factor kappa-B (NF-κB) is a key event in the release of inflammatory mediators [35]. Activation of the NF-κB signaling pathway induces the translocation of p65 to the nucleus, where it modulates the transcription of genes associated with inflammation [36]. p65 is predominantly located in the cytosol in unstimulated macrophages (Fig. 3H). However, p65 accumulates in the nucleus upon stimulation with LPS. Following Dex treatment, p65 nuclear translocation was reduced, yet a portion of it still entered the nucleus.
It was demonstrated that Dex@LIP and Dex@L-AB significantly inhibited p65 nuclear translocation, with Dex@L-AB exhibiting a more pronounced inhibitory effect than Dex@LIP. The reason for this may be that macrophages can take up more drugs through nanoparticles, and Dex@L-AB have a higher uptake efficiency and thus the best efficacy. In summary, the Dex@L-AB may exert multifarious anti-inflammatory effects by inhibiting the NF-κB signaling pathway, reducing NO, inflammatory cytokines, and scavenging ROS.
Ex Vivo biodistribution of L-AB
During apoptosis, PS acts as a key "eat-me" signal, exposing itself on the surface of apoptotic cells and AB [12]. This promotes the specific recognition by macrophages and subsequent internalization of the corpse [12]. Therefore, by taking advantage of the high affinity of macrophages for PS, L-AB were constructed for targeted drug delivery to macrophages. To track the in vivo distribution of AB, LIP, and L-AB, septic or healthy mice were intravenously injected with fluorescently labeled AB, LIP, or L-AB. The in vivo imaging system demonstrated that systemically administered AB were mainly localized in the liver and spleen of septic mice, which are the major organs of the mononuclear phagocyte system, consistent with previously reported studies [37]. It was demonstrated that nanoparticles injected via the tail vein could accumulate in the lungs of septic mice, likely due to endothelial damage and the resultant hyperpermeability [38]. Consequently, the increase in lung permeability induced the accumulation of LIP and L-AB in the lungs. L-AB exhibited a more pronounced accumulation and retention effect in the lungs of septic mice, with a fluorescence intensity approximately 1.6-fold greater than that of LIP at 4 h (p < 0.001). In addition, the distribution of L-AB in the liver, spleen, and kidneys was greater than that of LIP within the first four hours (Fig. 4A-B). This may be attributed to the "eat me" signal on the surface of L-AB.
In the initial 4 h following the onset of sepsis, the distribution of L-AB in the liver, spleen, and kidneys of septic mice was found to be significantly higher than that observed in healthy mice. The distribution of L-AB in the liver, spleen, and kidneys of healthy mice was found to be higher than that of septic mice at 12 h. This phenomenon may be attributed to the accumulation of immune cells, including macrophages, in the aforementioned organs, which facilitates the accelerated distribution of L-AB in inflammatory organs. Conversely, the modification of PEG prolonged the intracorporeal circulation of L-AB in mice, resulting in a slower distribution of L-AB to the liver and spleen of healthy mice. Furthermore, it was observed that the brightness of certain tissues exhibited a notable increase at 4 h compared to 1 h. However, the mean fluorescence signal of tissues at 4 h was found to be lower than at 1 h. One potential explanation for this phenomenon is that as inflammation progresses, there is a rise in the presence of inflammatory cells and tissue swelling, resulting in an increase in tissue mass (Fig. S5).
Furthermore, an analysis was conducted on the fluorescence intensity in blood, revealing that the distribution of AB was lower compared to that of LIP and L-AB (Fig. S6). This disparity may be attributed to the abundance of "eat me" signals present on the surface of AB, leading to their efficient elimination by the circulatory system. It is noteworthy that the average fluorescence intensity of L-AB in the bloodstream of septic mice was found to be similar to that of LIP at both 4 and 12 h post intravenous administration, suggesting that L-AB exhibited a prolonged circulation capacity akin to that of LIP.
To further confirm the uptake of L-AB by macrophages in various organs of mice, DiD-labeled AB, LIP, and L-AB were injected into septic or healthy mice via the tail vein. Subsequently, the co-localization of L-AB with the macrophage marker F4/80 in inflamed tissues was observed by CLSM (Fig. 4C-D, S7). The results demonstrated that the expression of F4/80 was elevated in the tissues of septic mice, and L-AB exhibited enhanced co-localization with F4/80 in various inflammatory tissues, suggesting that following the distribution of L-AB to various organs, they are primarily taken up by macrophages. It was observed that the distribution of AB in the lungs is uneven, with a few macrophages accumulating a large amount of AB around them. This may be related to the rapid uptake of AB by macrophages after it reached the lungs and cannot further spread. Additionally, the larger size of AB may also be a factor affecting its spread. The data indicate that L-AB possess the capacity to combine the benefits of long-term circulation of LIP and macrophage targeting of AB. This enabled them to enter major organs following systemic injection and be taken up by macrophages, which could be advantageous for the treatment of sepsis.
Anti-inflammatory therapeutic activity of Dex@L-AB in septic mice
Next, the therapeutic efficacy of Dex@L-AB was evaluated in a mouse model of LPS-induced sepsis (Fig. 5A). Mice were administered intravenously at 1 h after the establishment of the sepsis model with PBS, AB, free Dex, Dex@LIP or Dex@L-AB at a dosage of 1.5 mg/kg Dex. It was observed that all mice in the LPS group succumbed within 36 h (Fig. 5B). Although treatment with Dex resulted in an extension of survival time in some mice, the overall survival rate remained unchanged. The survival rate of mice in the Dex@LIP group increased to 50%, which may be attributed to the prolonged circulation and tissue retention time of LIP. After 144 h, only one mouse treated with AB survived (12.5%), whereas more than half of the mice treated with Dex@L-AB survived (75%), indicating that Dex@L-AB exerted a protective effect in the sepsis model. Furthermore, the cytokine storm in mice was evaluated by measuring the serum levels of inflammatory cytokines. Furthermore, the cytokine storm in mice was evaluated by measuring the serum levels of inflammatory cytokines. The cytokine levels in septic mice were found to be significantly higher than those in healthy mice. Both free Dex and Dex@LIP could reduce the level of proinflammatory cytokines to a certain extent. Dex@L-AB inhibited IL-6, MCP-1 and IL-12p70 levels more significantly in septic mice compared to Dex, while there was no significant difference in the ability to inhibit the levels of IFN-γ and TNF-α. The average level of those cytokines in Dex@L-AB treated group was lower than that in Dex@LIP treated group, suggesting that Dex@L-AB might have slender advantage in anti-inflammatory therapy (Fig. 5C-F, S8). Of these, Dex@L-AB was found to have the most significant treatment effect. Sepsis frequently results in acute impairment of the kidneys and liver, which can be evaluated via blood biochemistry and histological analysis (Fig. 5G-J). The administration of LPS notably elevated the levels of hepatic (aspartate transaminase [AST] and alanine transaminase [ALT]) and renal (blood urea nitrogen [BUN] and creatinine [CRE]) dysfunction markers in comparison to control mice. The administration of Dex@L-AB resulted in a significant improvement in the levels of these markers, indicating its efficacy in mitigating liver and kidney damage in septic mice.
Dex@L-AB attenuated the macrophage hyperactivation in vivo
Acute lung injury (ALI) is a common complication of sepsis. The pathology of ALI is characterized by the overactivation of macrophages and infiltration of immune cells in lung tissues [39]. To ascertain whether Dex@L-AB could suppress macrophage hyperactivation in septic mice, flow cytometry was employed to analyze the immune cells in lung tissues. As shown in Fig. 6A-B, Dex@L-AB exhibited a reduction in the CD11b+ F4/80+ macrophage population in septic mice, indicating that Dex@L-AB can effectively suppress macrophage activation and consequently inflammation. It is well established that M2 macrophages play a pivotal role in the resolution of inflammation and the repair of damaged tissues. The present study further investigated the potential of Dex@L-AB to promote the polarization of activated macrophages toward the M2 phenotype in vivo. As shown in Fig. 6C-D, the proportion of M1 macrophages (CD86+) was elevated (Fig. S9) and the proportion of M2 macrophages (CD206+) was diminished in the lungs of septic mice in comparison to healthy mice. Conversely, Dex@L-AB facilitated the transformation of macrophages into M2 macrophages. Furthermore, immunofluorescence staining identified pro-inflammatory factor iNOS and anti-inflammatory factor Arg-1 secreted by lung cells. It was observed that Dex@L-AB decreased the secretion of iNOS and increased the expression of Arg-1, indicating that the microenvironment of inflammation might be improved by Dex@L-AB (Fig. 6E). As shown in Fig. 6F, histological examination of lung tissue sections revealed that mice in the LPS group exhibited increased neutrophil infiltration, alveolar interstitial edema and thickened alveolar walls. Conversely, the different drug treatment groups showed notable reductions in these pathological manifestations, with Dex@L-AB exhibiting the most optimal therapeutic effect. MPO is a leukocyte enzyme released by activated neutrophils and monocytes, which forms free radicals and oxidative substances to kill pathogens [40]. However, it also results in oxidative damage to host tissues at the inflammatory sites [40]. There was an increase in MPO levels in the lung tissue following LPS stimulation. Nevertheless, MPO levels were significantly reduced following treatment with Dex@L-AB. In summary, Dex@L-AB inhibited the excessive activation of pulmonary macrophages in septic mice, thereby suppressing the inflammatory cytokine storm and reducing lung tissue damage.
Dex@L-AB attenuated cytokine storm-associated multiple organ damage in vivo
In the event of an infection-induced cytokine storm, multiple organ damage can occur, including acute liver and kidney injury in addition to pulmonary damage [2]. Therefore, it was also investigated whether treatment with Dex@L-AB could protect a number of organs from inflammatory damage in vivo. Following LPS challenge, it was observed that several organs exhibited clear pathological lesions, including hemorrhage and inflammatory cell infiltration. These lesions were observed in the liver, kidney, and spleen. Furthermore, liver necrosis, necrotic shedding of renal tubular epithelial cells to form casts and disorganized germinal centers, were also observed in mice challenged with LPS (Fig. 7A). In contrast, treatment with Dex@L-AB was found to suppress lesion formation in multiple organs. Excessive cytokine production can induce cell death by triggering the overproduction of ROS and subsequent activation of pro-apoptotic signals, ultimately accelerating the onset and progression of organ failure [41]. Furthermore, cellular apoptosis was monitored through immunofluorescence in various tissue sections. As shown in Fig. 7B, the number of apoptotic cells in the major organs (lung, liver and spleen) of LPS-treated mice was significantly elevated in comparison to the control group. However, this elevation was ameliorated by treatment with Dex@L-AB. In conclusion, Dex@L-AB demonstrated remarkable efficacy in the treatment of sepsis-induced organ injury by capitalizing on the advantages of prolonged circulation, macrophage targeting and the synergistic effects of dexamethasone and AB.
{kind=link}