Loss of inflammasome signaling does not prevent pro-inflammatory cytokine release and disease following SARS-CoV-2 infection in vivo.
To investigate the role of inflammasome pathways during severe SARS-CoV-2 infection we used a previously characterised mouse adapted strain (P21) 25. This strain was derived from a clinical isolate that was passaged 21 times in mice to allow adaptations that caused severe disease in WT mice, characterized by weight loss and increased levels of pro-inflammatory cytokines 25. During SARS-CoV-2 infection, activation of the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome pathway is thought to contribute to the release of bioactive IL-1β and IL-18, cytokines associated with severe COVID-19 5, 6, 26. We infected NLRP3 deficient mice (Nlrp3−/−) with P21 to explore this hypothesis. Interestingly, three days post infection (dpi), Nlrp3−/− mice displayed similar viral burdens to WT controls and did not show changes in weight loss, an established marker of disease severity in P21-infected mice at this time point (Fig. 1A-B). NLRP1 is also a potent activator of inflammation 27, 28, however, we observed that severe disease and lung viral burdens caused by SARS-CoV-2 infection were not affected by deletion of NLRP1 in mice (Fig. 1C-D). Many of the inflammasomes, including NLRP1, NLRP3, AIM2 and others, are dependent on the adapter protein ASC 13, 29, 30, 31. Surprisingly, ASC-deficient mice (Asc−/−) exhibited viral burdens and weight loss similar to infected WT animals (Fig. 1E-F). This demonstrates that ASC-dependent inflammasomes are not essential for SARS-CoV-2 driven pathogenesis.
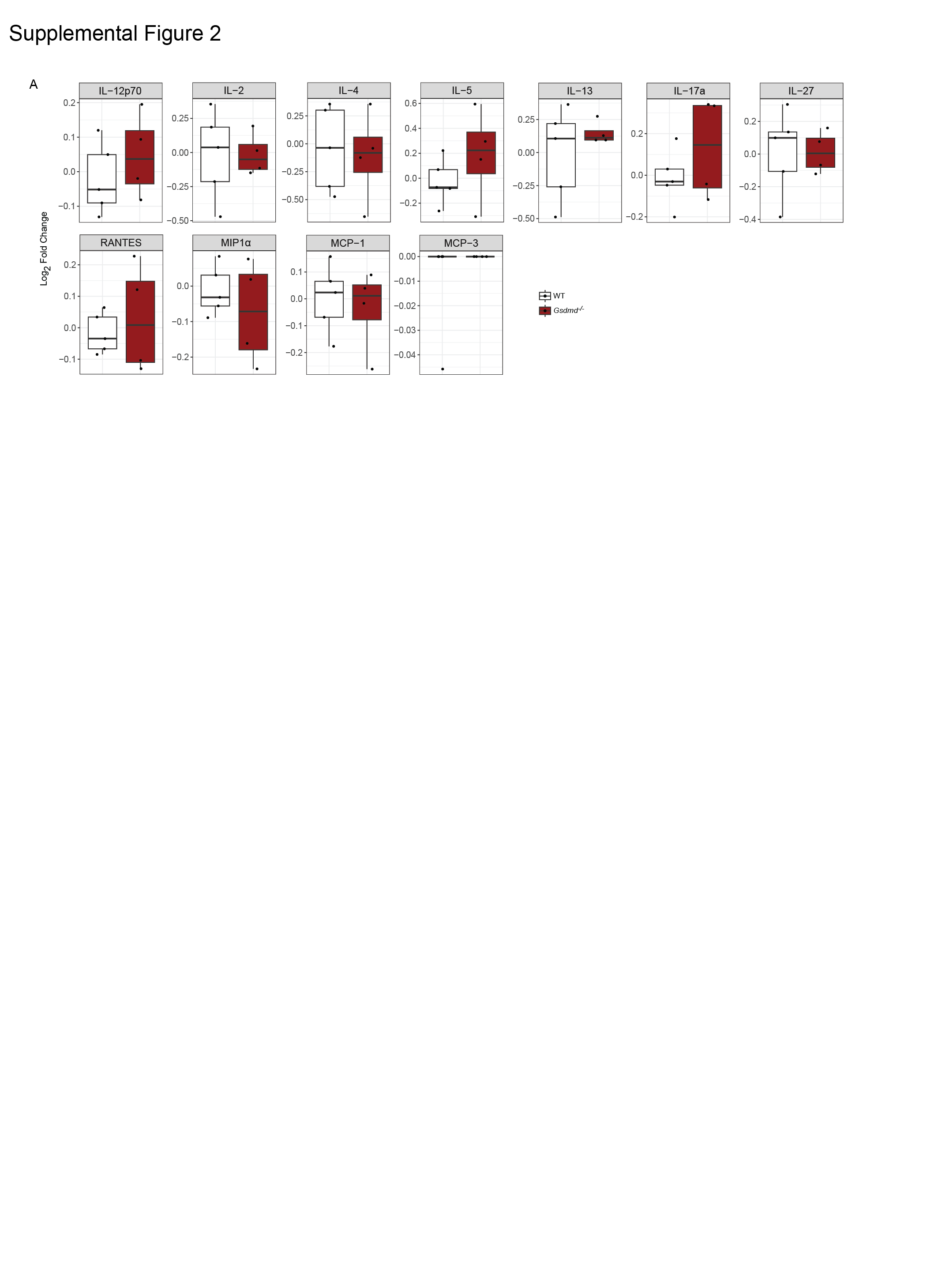
We next determined the abundance of 25 cytokines and chemokines in lung homogenates from infected gene-targeted mice to understand if lytic programmed cell death and canonical inflammasome activation contributed in a more subtle way to SARS-CoV-2 disease pathogenesis. Interestingly, neither of the inflammasome knockout mice we investigated displayed a reduction in IL-18, while only Asc−/− animals had slightly diminished IL-1β levels in their lungs upon SARS-CoV-2 infection (Fig. 1G). While Nlrp3−/−, Nlrp1−/− and Asc−/− inflammasome knockout mice all showed a reduction in the levels of TNF, MIP1α and IL-17a, variable differences in the levels of other cytokines were observed depending on the particular inflammasome deficiency (Fig. 1G and S1A). Overall, these results suggest that while NLRP1/3 and ASC do affect cytokine release during SARS-CoV-2 infection, this was not sufficient to impact disease severity (weight loss) or viral burdens in vivo compared to WT mice.
To better understand disease in SARS-CoV-2 P21 infected gene-targeted animals, we compared lung histology of knockouts and WT mice at 3 dpi. Lung sections were stained with hematoxylin and eosin (H&E) and analyzed by a board-certified pathologist. At 3 dpi, WT mice displayed multifocal, acute alveolitis (sometimes necrotizing), multifocal pneumonia, as well as moderate to severe acute multifocal perivasculitis (Fig. 1H). Gene-targeted mice showed similar manifestations of disease with interstitial pneumonia, moderate to severe multifocal perivasculitis and acute alveolitis. Immunohistochemical (IHC) staining for SARS-CoV-2 nucleocapsid showed that the virus localized to the bronchiolar and alveolar epithelium, as well as macrophages in both WT and all gene-targeted mice (Fig. 1H). Staining for myeloperoxidase (MPO), CD3 and F4/80 revealed a similar number of myeloid cell and T cell infiltrates in WT and all knockout mice (Fig. 1H). Collectively, these findings show that while canonical ASC-dependent inflammasome pathways do play some role in the cytokine responses associated with severe SARS-CoV-2 pathogenesis, they do not significantly impact disease outcomes.
The pyroptosis effector Gasdermin D is not required for pro-inflammatory cytokine release during SARS-CoV-2 infection in vivo.
Regardless of the upstream mechanisms responsible for the activation of pyroptosis and cytokine production, the cellular release of cytokines and cell lysis is thought to be primarily dependent on the activation of the pore forming protein GSDMD 32. We found that GSDMD deficient animals (Gsdmd−/−) showed similar viral burdens and weight loss compared to WT mice upon infection with SARS-CoV-2 (Fig. 2A-B). Analysis of 25 cytokines and chemokines in lung homogenates from infected Gsdmd−/− mice showed that IL-1β levels were slightly reduced compared to WT animals (Fig. 2C). Interestingly, however, the levels of IL-18 and IL-23 were increased in these animals upon infection (Fig. 2C and S2A). These data indicate that GSDMD does not exert an essential role in the pathogenic pro-inflammatory cytokine release during SARS-CoV-2 infection.
We further compared lung histology of Gsdmd−/− and WT mice at 3 dpi. Lung sections were stained with hematoxylin and eosin (H&E) and analyzed by a board-certified pathologist. Three days post SARS-CoV-2 infection, both Gsdmd−/− and WT mice displayed multifocal, acute alveolitis, multifocal pneumonia, as well as moderate to severe acute multifocal perivasculitis. IHC staining for SARS-CoV-2 nucleocapsid showed that virus localized to the bronchiolar and alveolar epithelium in both WT and Gsdmd−/− mice. MPO, CD3 and F4/80 staining revealed a similar pattern of myeloid cell and T cell infiltrates in WT and Gsdmd−/− animals (Fig. 2D).
We next explored if severe SARS-CoV-2 disease was linked to cleavage (i.e. activation) of GSDMD in WT animals. In line with our previous results, no cleaved GSDMD was detected by IHC in WT animals with severe SARS-CoV-2 disease confirmed with nucleocapsid staining (Fig. 2E). As a positive control, to ensure that our antibody detects cleaved GSDMD, we stained small intestine tissue from mice infected with the enteric pathogen Cryptosporidium, which is known to trigger pyroptosis and cause Gasdermin D cleavage 33 (Fig. 2E bottom panel). To further investigate GSDMD cleavage during SARS-CoV-2 infection, we performed Western blot analysis of whole lung tissue extracts. Cleavage (i.e. activation) of GSDMD in the lungs of infected animals could not be detected in WT and in Asc−/− mice (Fig. 2F, original westerns can be found in the supplemental information). Together with our genetic investigations, this indicates that the inflammation associated with severe SARS-CoV-2 disease does not lead to or require the activation of GSDMD in vivo.
Regulators of non-canonical pyroptosis are dispensable for SARS-CoV-2 infection induced pathogenesis.
We further investigated the role of non-canonical pyroptosis pathways, including those involving GSDME activation. We infected Gsdme−/− mice with SARS-CoV-2 but found no difference in disease phenotype compared to control WT animals. Furthermore, compound loss of GSDMD/E (Gsdmd/e−/−) and GSDMA/C/E (Gsdma/c/e−/−) did not alter disease phenotypes compared to SARS-CoV-2 infected WT mice (Fig. 3A-D).
Functional overlap of programmed cell death pathways, particularly in response to infection, has emerged as a paradigm in recent years 34, 35. To examine if necroptosis could be compensating for the loss of pyroptosis, we infected mice lacking essential effectors of both of these lytic programmed (regulated) cell death processes, namely GSDMD and MLKL, with SARS-CoV-2. Compound mutant Gsdmd/Mlkl−/− mice showed similar disease phenotypes compared to infected WT animals (Fig. 3E-F). We extended our investigation to examine mice that were deficient in NINJ1, a protein that drives plasma membrane rupture and release of high molecular weight DAMP downstream of pyroptosis, apoptosis, ferroptosis and accidental cell lysis, but not necroptosis 36, 37. Ninj1−/− mice showed similar disease phenotypes upon SARS-CoV-2 infection as WT mice (Fig. 3G-H).
Cytokine processing during SARS-CoV-2 infection occurs independently of caspases-1/-11/-12.
Inflammasome and pyroptosis signaling have been implicated in the pathogenesis of COVID-19 disease because of their known link to IL-1β and IL-18 release. Both of these cytokines have been associated with increased severity of this disease 4, 38, 39. The prevailing dogma is that these cytokines are released downstream of inflammasome signaling and processed into their bioactive forms by caspases-1, -11 (and possibly − 12) 40, 41, 42. We therefore used gene-targeted mice lacking all these caspases (C1/11/12−/−) to understand their overall contribution to severe SARS-CoV-2 disease in our mouse model. Infected C1/11/12−/− mice showed similar viral burdens and weight loss compared to infected WT mice (Fig. 4A-B). Analysis of 25 cytokines and chemokines showed that upon SARS-CoV-2 infection, C1/11/12−/− mice mounted a similar cytokine/chemokine response to infected WT controls, with only GROα being slightly elevated in knockout mice (Fig. 4C and S4A). This finding indicates that processing and release of pro-inflammatory IL-1β and IL-18 during SARS-CoV-2 infection can occur independently of the three inflammatory caspases-1/-11/-12. Histological examination confirmed similar lung pathology and immune cell infiltrates in SARS-CoV-2 infected C1/11/12−/− and WT mice (Fig. 4D). These results show that all components of the pyroptosis machinery, from ASC-dependent inflammasome sensors to catalytic pro-inflammatory caspases and the pore-forming effector proteins, are not essential to drive SARS-CoV-2 viremia or disease manifestations.
IL1-b, but not IL-18 contributes to severe SARS-CoV-2-mediated disease.
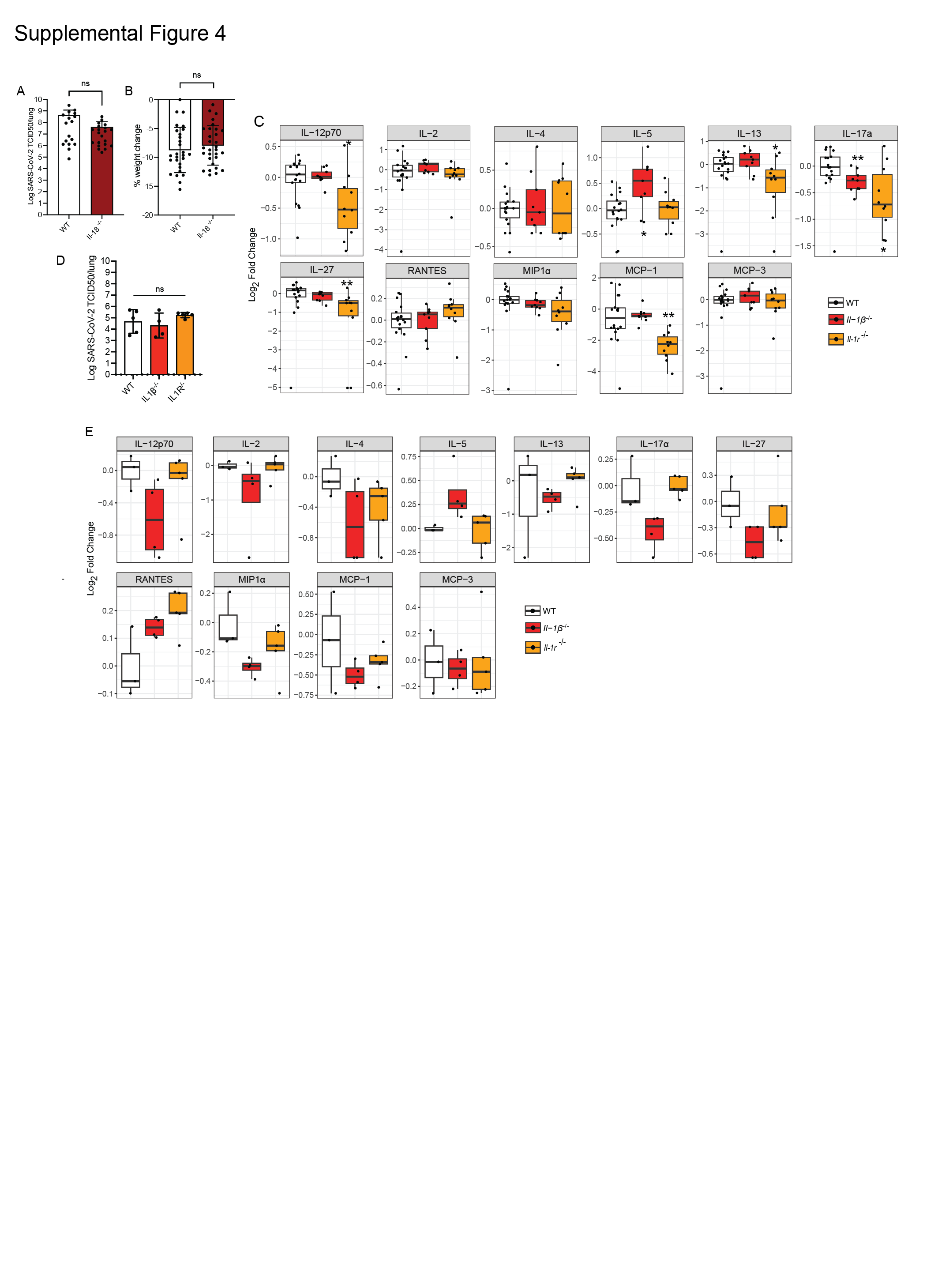
Our results show that SARS-CoV-2 driven inflammation and cytokine release in vivo are independent of key components of the pyroptosis machinery. Several studies reported an association between IL-1β and IL-18 serum levels and severe COVID-19 disease 5, 6, 26. Pyroptosis and caspases-1/-11/-12 are often linked to IL-1β and IL-18 processing. So, if these inflammatory cytokines are critically involved in severe COVID-19 disease, the results from our studies using gene targeted mice indicate a disconnect between the processing and release of IL-1β and IL-18 and pyroptosis. To confirm that one or both of these cytokines do contribute to SARS-CoV-2 driven disease in our animal model, we infected IL-18 deficient (Il-18−/−) mice. Il-18−/− mice were not significantly protected from SARS-CoV-2 infection displaying similar disease compared to infected WT animals (Fig. S5A-B). In contrast, Il-1β−/− mice had significantly lower viral burdens in the lungs, and exhibited less weight loss compared to infected WT animals (Fig. 5A-B).
To better understand the cellular host responses linked to better outcomes in SARS-CoV-2 infected Il-1β−/− mice, we examined lung histology at 3 dpi. Lung sections were stained with hematoxylin and eosin (H&E) and analyzed by a board-certified pathologist. Three days post SARS-CoV-2 infection, WT mice had mild to severe acute multifocal perivasculitis, interstitial pneumonia and necrotizing alveolitis. Infected IL-1β deficient mice also showed perivasculitis, moderate interstitial pneumonia, but no alveolitis (Fig. 5C-D). IHC staining for SARS-CoV-2 nucleocapsid showed that virus localized to the bronchiolar and alveolar epithelium and macrophages in both WT and Il-1β−/− mice. Histological scoring showed that in contrast to WT controls, Il-1β−/− mice did not present with signs of pleural mesothelial hyperplasia, iBALT or proteinaceous debris in the air space. These differences amounted to an overall significantly reduced histological score of disease (Fig. 5C-D). These findings in gene-targeted mice corroborate the human correlative data and prove for the first time in an in vivo setting that IL-1β plays a critical pathogenic role during severe SARS-CoV-2 induced disease.
Inflammatory IL-1 signaling is activated through binding of IL-1 family cytokines to the membrane bound, type I interleukin-1 receptor (IL-1R) 43. Interestingly, while Il-1r−/− animals were also protected from severe SARS-CoV-2 induced disease, as measured by a reduction in weight loss, these animals did not have decreased viral burdens but were similar to WT mice (Fig. 5E-F). To identify the effects of the absence of critical components of the IL-1 pathway on the pro-inflammatory cytokine response, we quantified the levels of 25 cytokines and chemokines in the lungs of WT, Il-1β−/− and Il-1r−/− animals at 3 dpi. The absence of IL-1β led to reductions in GM-CSF, IL17a and IL-5. Il-1r−/− animals tended to express lower levels of a wider range of cytokines and chemokines compared to IL-1β deficient mice (Fig. 5G and S5C). However, the reduction in cytokines was of longer duration, continuing for 6 days post-infection, and was more profound at later time points in IL-1β deficient mice compared to WT animals and Il-1r−/− animals (Fig. 5H and S5D).
Age related severity of SARS-CoV-2 disease can be reduced by the absence of IL1-b.
Similar to COVID-19 in humans, SARS-CoV-2 P21 driven disease in mice becomes more pronounced with increased age 25. Upon infection, the majority of infected WT animals > 10 weeks-old lose more than 20% body weight, reaching ethical end-point by 4–6 dpi 25. To test whether the absence of IL-1β could protect against aged-associated severity of SARS-CoV-2 driven disease, we infected eleven-week-old Il-1β−/− and control WT mice. IL-1β deficient mice were more likely to survive SARS-CoV-2 infection compared to WT controls (Fig. 6A), however, this survival advantage was attenuated in 6-month-old infected mice (Fig. 6B). Despite this, 6-month-old IL-1β deficient mice showed significantly lower viral burdens in the lungs at the peak of infection (3 dpi) compared to WT controls (Fig. 6C-D). Interestingly, in aged animals (> 6 months), while multiple cytokines were decreased at 3 dpi, the only cytokine found to be significantly reduced in lung homogenates from Il-1β−/− mice was IL-1β itself (Fig. 6E). To analyse the potential of anti-IL-1β therapeutics against severe COVID-19 in our mouse model, we administered a single dose of IL-1β neutralizing monoclonal antibodies to 10- to 12-week-old WT mice on the day of infection. Inhibition of IL-1β alone was not sufficient to prevent mortality in P21 infected animals although a trend was observed. Thus, more profound inhibition of IL-1β and concomitant inhibition of other pathogenic cytokines should be explored.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}