SADS-CoV infection leads to changes in the structure of the microbiota
High-throughput sequencing of the 16S rRNA gene was conducted to explore the differences in the microbiota composition of the ileum mucosa and feces between healthy and SADS-CoV-infected piglets. On average, 76,676 sequences per sample were generated (range: 53,073–96,405) for the 16S rRNA gene V4 region, with an average effective tag length of 253 bp.
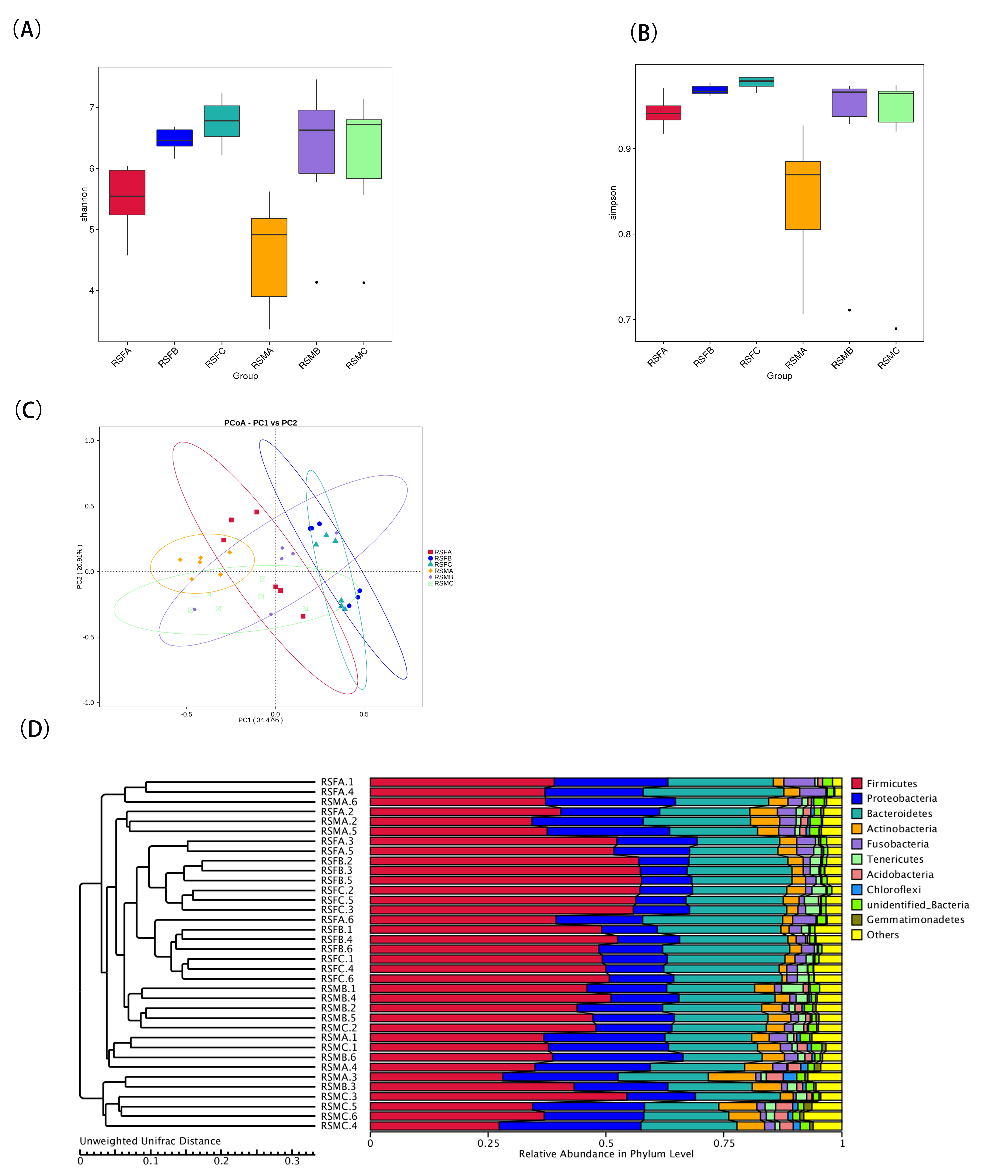
The dominant bacterial composition in the ileum mucosa and feces of piglets was altered by SADS-CoV infection across various taxonomic levels (Supplementary Fig. 1). Furthermore, the Shannon and Simpson indices were greater in piglets in the control group and SADS-CoV P83 group than in those in the SADS-CoV P7group (Supplementary Figs. 2A and 2B).
Beta diversity among the groups was assessed via principal coordinate analysis (PCoA) based on weighted UniFrac distances. The resulting scatterplot revealed six distinct clusters representing gut bacterial communities (Supplementary Fig. 2C). Notably, samples from the SADS-CoV P7-infected group and the control group tended to have different clusters, whereas samples from the SADS-CoV P83-infected group and the control group displayed substantial overlap, suggesting a degree of similarity in microbial composition between these two groups.
To further elucidate the similarity among samples, a clustering tree using the unweighted pair-group method with arithmetic mean (UPGMA) based on weighted UniFrac distances was constructed (Supplementary Fig. 2D). The clustering tree effectively differentiated the SADS-CoV P7-infected samples from the control group samples in both the ileum mucosa and feces of piglets.
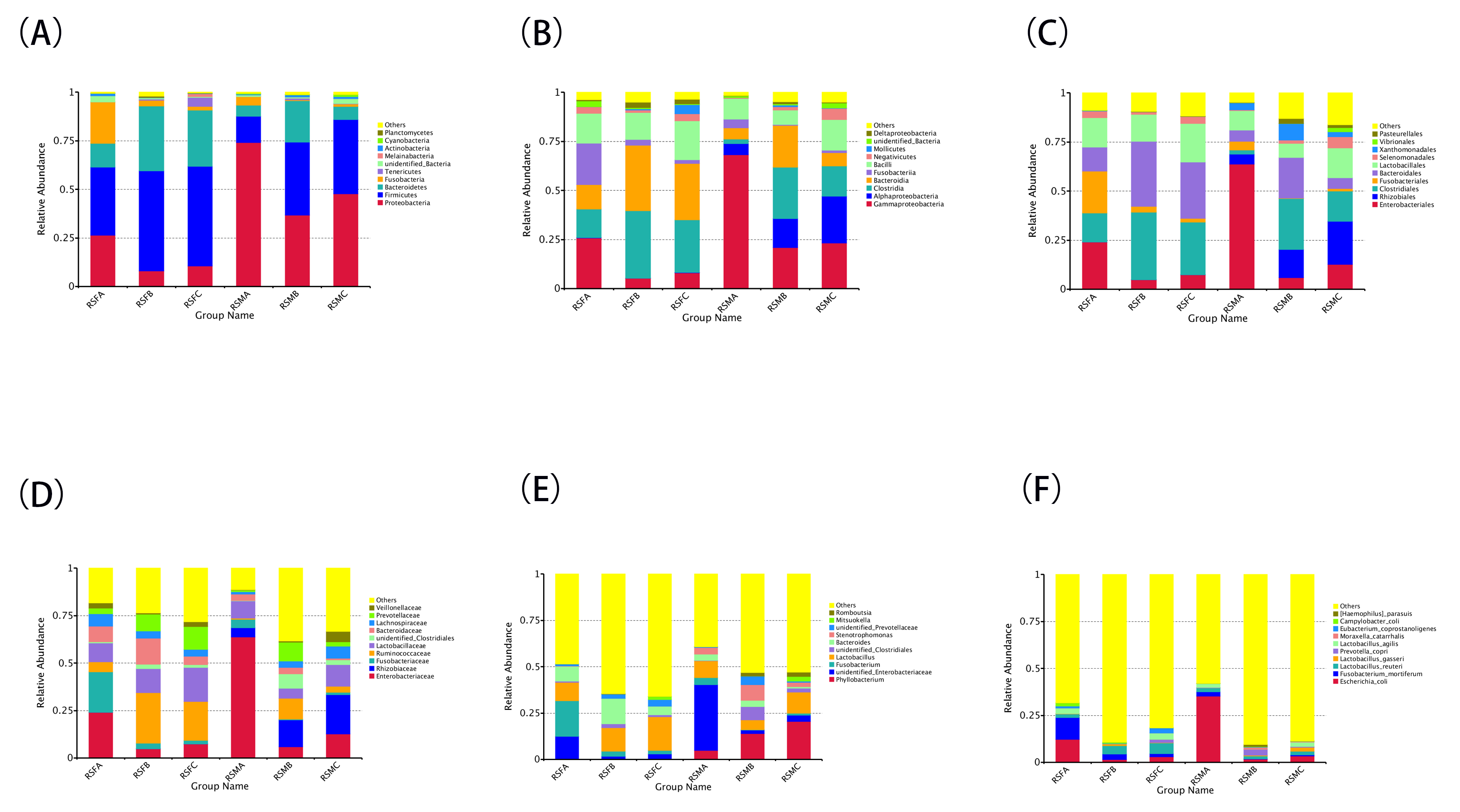
To delve deeper into the differences in microbiota distribution, we conducted MetaStat analyses focusing on the top 35 genera across the three groups. As illustrated in Fig. 2A, following SADS-CoV P7 infection, there was a notable increase in the abundance of pathogenic bacteria associated with diseases that cause diarrhea. In particular, the proportion of unidentified Enterobacteriaceae in the ileum mucosa and feces of the SADS-CoV P7-infected group was markedly high (35.48% and 12.50%, respectively), whereas the proportion was substantially lower in the SADS-CoV P83-infected group and the control group, both in the ileum mucosa (1.77% and 1.58%, respectively) and feces (3.43% and 2.93%, respectively). Furthermore, the proportions of Fusobacterium and Megasphaera in the feces of the SADS-CoV P7-infected group were significantly greater than those in the feces of the SADS-CoV P83-infected group (19.10% and 1.35% vs. 2.90% and 0.01%, respectively) and the control group (1.95% and 0.16%, respectively). Additionally, the proportion of Campylobacter in the feces of the SADS-CoV P7-infected group (1.63%) was significantly greater than that in the feces of the control group (0.02%).
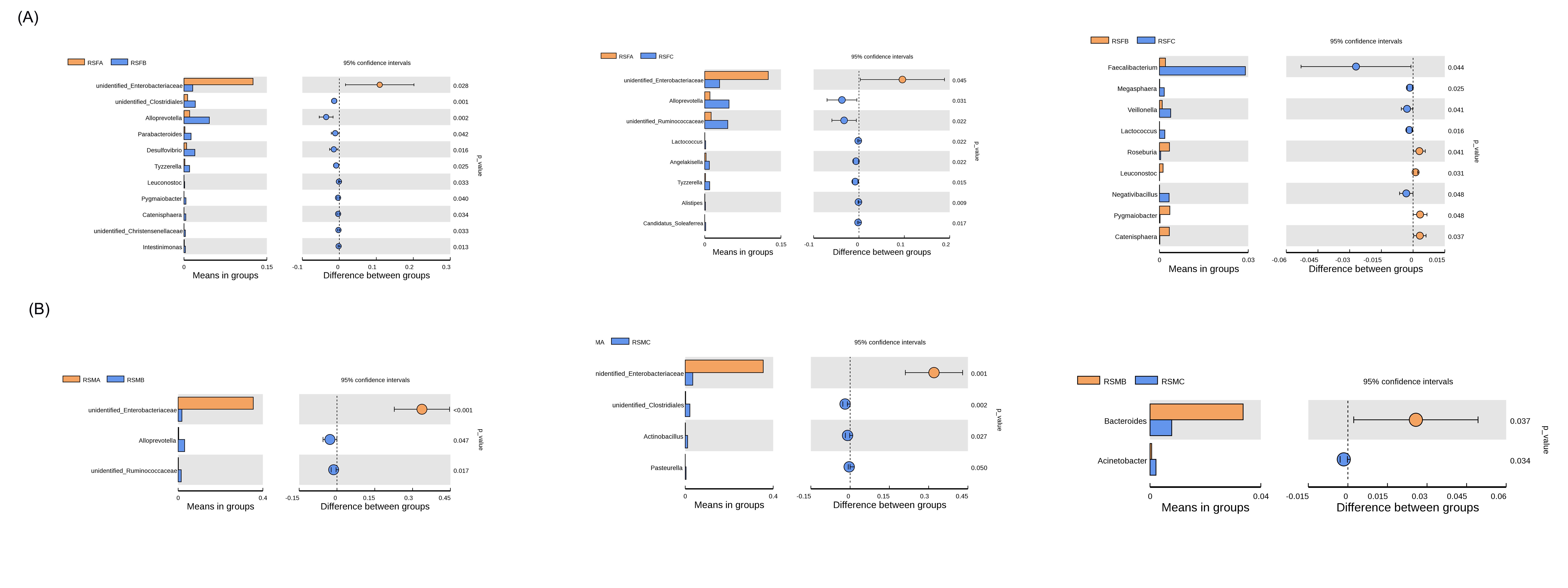
Conversely, there was a decrease in the abundance of bacteria beneficial for intestinal health in the SADS-CoV P7-infected group. The proportions of Faecalibacterium, unidentified Ruminococcaceae, Actinobacillus, and Streptococcus in the ileum mucosa of the SADS-CoV P7-infected group were significantly lower than those in the ileum mucosa of the SADS-CoV P83-infected group and the control group. Similarly, the proportions of Turicibacter and Alloprevotella in the feces of the SADS-CoV P7-infected group were significantly lower than those in the feces of the SADS-CoV P83-infected group and the control group. Moreover, the proportions of Faecalibacterium and unidentified Ruminococcaceae in the feces of both the SADS-CoV P7-infected and SADS-CoV P83-infected groups were significantly lower than those in the control group. Furthermore, the proportion of unidentified Clostridiales in the ileum mucosa of the SADS-CoV P7-infected group was significantly lower than that in the ileum mucosa of the control group. Similarly, the proportion of Terrisporobacter in both the ileum mucosa and feces of the SADS-CoV P7-infected group was significantly lower than that in the control group. Notably, Comamonas could no longer be detected in either the ileum mucosa or feces of the SADS-CoV P7-infected group, whereas in the control group, there was a low but identifiable proportion (0.78% and 0.0004%, respectively).
Interestingly, the proportions of unidentified Prevotellaceae, Alloprevotella, Parabacteroides, Faecalibacterium, and unidentified Ruminococcaceae in the ileum mucosa, as well as Turicibacter in the feces of the SADS-CoV P83-infected group, were significantly greater than those in the SADS-CoV P7-infected group and the control group. Additionally, the proportions of unidentified Lachnospiraceae, Romboutsia, Collinsella, and Subdoligranulum in the ileum mucosa of the SADS-CoV P83-infected group were significantly greater than those in the ileum mucosa of the SADS-CoV P7-infected group (Fig. 2B). Detailed interspecies differences at the genus level are shown in the Supplementary Fig. 3.
Using LEfSe analysis, we evaluated the significant differences in microbial abundance in the ileum mucosa (Fig. 3A and B) and feces (Fig. 3C and D) of suckling piglets from the phylum level to the species level and plotted cladograms representing the structure of the host-microbiota axis. As shown in Fig. 3A and 3B, the cladogram indicated significant shifts in each of the three groups. Among the three groups, 24 phylotypes from phylum to species were identified as high-dimensional biomarkers. These microbes mainly belonged to six different phyla, namely, Firmicutes, Bacteroidetes, and Proteobacteria. The abundance of E. coli, which belongs to the phylum Proteobacteria, was significantly greater in the RSMA group. The abundance of the phyla Firmicutes and Proteobacteria in the RSMB group substantially increased due to an increase in the abundance of the order unidentified Clostridiales; family Ruminococcaceae, belonging to the phylum Firmicutes; and unidentified genera of the family Prevotellaceae, belonging to the phylum Proteobacteria.
The RSMC group had distinct mucosal microbial compositions mostly belonging to the phylum Firmicutes, represented by the genus Mitsuokella, which belongs to the class Negativicutes; the family Streptococcaceae; and the species Lactobacillus agilis, the latter two of which belong to the order Lactobacillales. As shown in Fig. 3C and 3D, the cladogram indicated significant shifts in each of the three groups. Among the three groups, 25 phylotypes from phylum to species were identified as high-dimensional biomarkers. These microbes mainly belonged to six different phyla, namely, Firmicutes, Bacteroidetes, Proteobacteria, Fusobacteria, Tenericutes, and unidentified bacteria. The abundance of the species E. coli; the genus Fusobacterium, belonging to the phylum Fusobacteria; the order Campylobacterales, belonging to the phylum “unidentified Bacteria”; and the genus Peptostreptococcus, belonging to the phylum Firmicutes, was significantly greater in the RSMA group. The abundance of the phyla Firmicutes and Proteobacteria in the RSMB group substantially increased due to an increase in the abundance of the order Clostridiales, which belongs to the phylum Firmicutes, and the classes Deltaproteobacteria and Gammaproteobacteria, which belong to the phylum Proteobacteria. The RSMC group had distinct fecal microbial compositions, mostly belonging to the phylum Bacteroidetes and phylum Firmicutes, represented by the family Muribaculaceae and genus Alloprevotella belonging to the phylum Bacteroidetes, and the species Lactobacillus agilis, species Eubacterium coprostanoligenes, and genus Faecalibacterium belonging to the Firmicutes.
Microorganisms in the ileum mucosa and feces were both selected as research objects. As shown in Fig. 3 (E)-(G), the microbial structures of the three groups were obviously different. In the control group, Lactobacillus gasseri and the genus Romboutsia, belonging to the Firmicutes, the genera Stenotrophomonas and Vibrio, and the order Rhizobiales, belonging to the Proteobacteria, were mainly distributed in the ileum mucosa; the species Eubacterium coprostanoligenes and the genus Faecalibacterium, both belonging to the phylum Firmicutes; the genus unidentified Prevotella, and the genus Alloprevotella and the family Muribaculaceae, belonging to the phylum Bacteroidetes; and the class Mollicutes, belonging to the phylum Tenericutes, were mainly distributed in the feces. In the SADS-CoV P83-infected group, the family Rhizobiaceae and genera Actinobacillus and Stenotrophomonas, all belonging to the phylum Proteobacteria; the species Prevotella copri (phylum Bacteroidetes), and the genus Romboutsia (phylum Firmicutes) were mainly distributed in the ileum mucosa; the genus Ruminococcaceae (phylum Firmicutes) was mainly distributed in the feces. In the SADS-CoV P7-infected group, the species E. coli and the family Rhizobiaceae, which belong to the phylum Proteobacteria, were mainly distributed in the ileum mucosa; the class Negativicutes, the order Selenomonadales, and the family unidentified_Lachnospiraceae, all belong to the phylum Firmicutes; the species Fusobacterium mortiferum (phylum Fusobacteria); and the order Campylobacterales (“unidentified Bacteria phylum”) were mainly distributed in the feces.
Microbial functions were predicted using Tax4Fun based on the relative abundance of microbes. Overall, 35 functional groups were identified in the microbiota of the ileum mucosa and feces. “Carbohydrate metabolism”, “membrane transport”, “replication and repair”, “translation” and “amino acid metabolism” were the top five functional annotations in the three groups, followed by “energy metabolism”, “nucleotide metabolism”, “glycan biosynthesis and metabolism”, “metabolism of cofactors and vitamins”, and “signal transduction” (Fig. 4A). After functional prediction of each group, cluster analysis was carried out using a heatmap. As shown in Fig. 4B, the mucosa and fecal microbial functional clusters in the SADS-CoV P7-infected group were quite different from those in the SADS-CoV P83-infected group and the control group. We found that the mucosa and fecal microbial functional clusters were more similar between the SADS-CoV P83-infected group and the control group, although their regions of relative abundance differed.
In summary, highly pathogenic SADS-CoV P7 increased the proportion of harmful bacteria and decreased the proportion of beneficial bacteria in the ileum mucosa and feces of piglets, while low-pathogenicity SADS-CoV P83 altered the microbial structure without obvious clinical manifestations. Notably, our results showed that the abundance of the opportunistic pathogen E. coli was significantly greater in the RSMA group than in the other groups.
Abnormal metabolism of short-chain fatty acids is accompanied by microbial dysbiosis in SADS-CoV-infected piglets
The short-chain fatty acids (SCFAs) in the contents of the large intestine were examined. As shown in Fig. 5A, the concentrations of SCFAs, including acetate, propionate, butyrate, isobutyrate, valerate, isovalerate, and hexanoic acid, in SADS-CoV P7-infected pigs were significantly lower than those in SADS-CoV 83-infected pigs (P < 0.01). The concentrations of acetate, propionate, butyrate, isobutyrate, valerate, and isovalerate were also significantly lower in SADS-CoV P7-infected pigs than in control group pigs (P < 0.01), whereas in the SADS-CoV P83-infected pigs, the concentrations of acetate, propionate, butyrate, and valerate were significantly lower than those in the control group pigs (P < 0.05), indicating some differences in SCFA content between the two groups of infected pigs.
Interestingly, the levels of acetate, propionate, butyrate, isobutyrate, valerate, and isovalerate were negatively correlated with the levels of Enterobacteriaceae and Escherichia coli in the ileum mucosa but were positively correlated with those of the family Clostridiales, genus Clostridiales, and family Peptostreptococcaceae in the ileum mucosa. Acetate, propionate, butyrate, isobutyrate, valerate, and isovalerate levels were negatively correlated with the levels of Fusobacteria, order Fusobacteriales, class Fusobacteriia, family Fusobacteriaceae, genus Fusobacterium, phylum unidentified_Bacteria, class unidentified_Bacteria, order Campylobacterales, and genus Peptostreptococcus in the feces but were positively correlated with the levels of the family Muribaculaceae, class Mollicutes, phylum Tenericutes, genus Alloprevotella, and family Ruminococcaceae in the feces (Fig. 5B).
Global overview of metabolism in the fecal metabolome
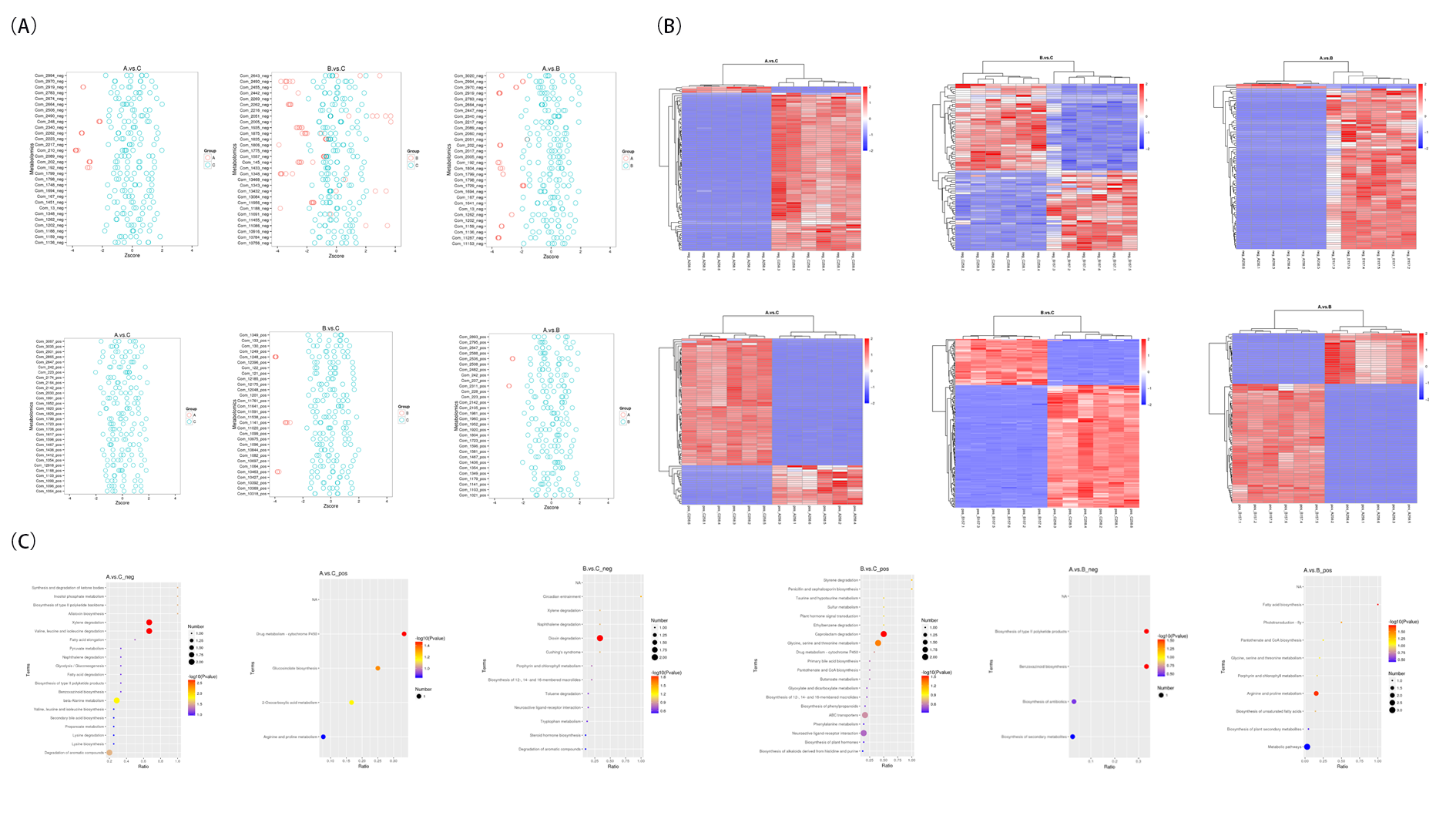
By utilizing variable importance in projection (VIP) scores derived from the first principal component of partial least squares-discriminant analysis (PLS-DA) model, in conjunction with T-test P values, we identified differentially abundant metabolites in the fecal metabolome, employing the criteria of a VIP > 2.0, a fold change (FC) exceeding 2.0 or less than 0.5, and P values below 0.05. This methodology revealed 157 differentially abundant metabolites between the SADS-CoV P7 infection group and the control group, comprising 76 cationic and 81 anionic metabolites. For the SADS-CoV P83 infection group versus the control group, 249 differentially abundant metabolites were detected, with 172 in cation mode and 77 in anion mode. Moreover, a comparison of the SADS-CoV P7 and P83 infection groups revealed 148 differentially expressed metabolites, which were evenly distributed, with 74 in both the cation and anion modes.
This investigation successfully identified differentially expressed metabolites across three comparisons: SADS-CoV P7 versus control, SADS-CoV P83 versus control, and SADS-CoV P7 versus SADS-CoV P83. Particularly, in the comparison between the SADS-CoV P7 group and the control group, cation mode analysis revealed notable upregulation of metabolites such as alfentanil, 2-O-sulfo-L-idopyranuronic acid, and creatinine, whereas ethamivan, toloxatone, and mepivacaine exhibited significant downregulation. Anion mode analysis revealed downregulation of metabolites such as 2,6-diamino-7-hydroxynonanedioic acid and 3a,7a-dihydroxycholanoic acid, with only a few, such as paracetamol sulfate, showing significant upregulation. In the comparison between SADS-CoV P83 and the control group, upregulated cationic metabolites included iscotrizinol and rebamipide, while significant downregulation was observed for metabolites such as ±-lauroylcarnitine. In the anionic mode, notably downregulated compounds included 2-[(dimethylamino)] and hexadecanedioic acid mono-L-carnitine ester, with azelaic acid among the few that were significantly upregulated. Among the SADS-CoV P7 and P83 groups, compared to the control group, the upregulated cationic metabolites included decanoylcarnitine, while the downregulated cationic metabolites included D-panthenol and mepivacaine. Anionic analysis revealed downregulation of prostaglandin A1 ethyl ester and upregulation of metabolites such as linalyl butyrate (Supplementary Fig. 4A).
Hierarchical clustering analysis of the primary differentially expressed metabolites across treatment groups revealed entirely divergent metabolic expression trends between the SADS-CoV P7 and SADS-CoV P83 groups, the SADS-CoV P7 and control groups, and the SADS-CoV 83 and control groups, underscoring the distinct metabolic impacts of each SADS-CoV infection scenario (Supplementary Fig. 4B).
Through enrichment analysis utilizing the KEGG pathway database, we investigated the metabolic pathways associated with differentially expressed metabolites among the SADS-CoV P83 and SADS-CoV P7 infection groups and the control group. As illustrated in Supplemental Fig. 4C, in cation mode, the metabolites that were differentially abundant between the SADS-CoV P7 group and the SADS-CoV P83 group were primarily associated with fatty acid biosynthesis and arginine and proline metabolism pathways; in the anionic mode, the differentially abundant metabolites were mainly linked to benzoxazinoid biosynthesis and the biosynthesis of type II polyketide products. Furthermore, in the comparison with the control group, the differentially abundant metabolites in the SADS-CoV P7 group were predominantly associated with valine, leucine, and isoleucine degradation; beta-alanine metabolism; and the synthesis and degradation of ketone bodies. In cation mode, the metabolites that were differentially abundant between the SADS-CoV P7 group and the control group were primarily related to the drug metabolism-cytochrome P450 and glucosinolate biosynthesis pathways. Additionally, in the cation mode, in the comparison with the control group, the differentially abundant metabolites in the SADS-CoV P83 group were mainly linked to caprolactam degradation and glycine, serine, and threonine metabolism pathways. In the anionic mode, the metabolites that were differentially abundant between the SADS-CoV P83 group and the control group were primarily associated with dioxin degradation and circadian entrainment pathways.
Analysis of the correlations between the microbiota and metabolites
We separately analyzed the NGS (Next-generation Sequencing) or mass spectrometry data of proteomics, metabolomics, short-chain fatty acid analysis, and 16S rRNA sequencing through basic bioinformatics analysis, expression/abundance detection, and differential expression analysis between samples. In this context, "A" represents the SADS-CoV P7 group, "B" represents the SADS-CoV P83 group, and "C" denotes the control group. The outcomes were further analyzed based on the intersections of A-vs-C and A-vs-B, as well as B-vs-C and A-vs-C. Consequently, we identified molecular events related to pathogenicity (P set) and immune responses (I-V set) in each of these groups. We considered the intersection of A-vs-C and A-vs-B to be the P set, and the intersection of B-vs-C and A-vs-C to be the I-V set.
Hierarchical all-against-all association (HAllA) analysis was performed to explore the correlation between gut bacteria and metabolites. Metabolite information is shown in Supplement Table 1. In the P set, in the feces, in the anion mode, indole-3-carbidol; sedanolide; lysoPC (18:4(6Z,9Z,12Z,15Z)); 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine; and 8,9a-dihydroxy-3,3b,5',5b-tetramethylicosahydro-3H-spiro[benzo[6, 7]-as-indaceno[2,3-b]furan-2,2'-pyran]-10-carbaldehyde were significantly negatively correlated with the genus unidentified_Fusobacteriaceae. Unoprostone, canthiumine, premithramycinone, and 3-methoxy-4-hydroxyhippuric acid were significantly positively correlated with the genus Alloprevotella. In the cation mode, norverapamil, toloxatone, and ethamivan were significantly negatively correlated with the genus Fusobacterium, and ethamivan, toloxatone, norverapamil, and 1-phenyl-1,3-octadecanedione were significantly positively correlated with the genus Alloprevotella (Fig. 6A). In the ileum mucosa, anionic 2,6-diamino-7-hydroxynonanedioic acid and trans-geranic acid were significantly positively correlated with the phylum Firmicutes (Fig. 6B). In cation mode, 2-O-sulfo-L-idopyranuronic acid was significantly negatively correlated with the phylum Firmicutes. R1128B, guaifenesin, and ethamivan were significantly negatively correlated with the genus unidentified_Enterobacteriaceae. Ethamans, n-acetylsphingosine, and toloxatone were positively correlated with the phylum Firmicutes (Fig. 6B). In I-V, in the feces, in the anion mode, unoprostone and methyl3-acetoxy-11-methoxyurs-12-en-28-oate were significantly negatively correlated with the genus Fusobacterium, and unoprostone and 2-(dimethylamino)-5,6-dimethylpyrimidin-4-ol were significantly positively correlated with the genus Alloprevotella. In the cation mode, (Z)-N-[(2R)-2-amino-1-hydroxy-3-{[(1E)-N-hydroxy-7-(methylsulfanyl)heptanimidoyl]sulfanyl}propylidene]glycine, 2-oxohex-4-enoic acid, and N-desmethyltramadol were significantly negatively correlated with the genus Fusobacterium. TDIQ, 2E-crotamiton, and prilocaine were significantly positively correlated with the genus Alloprevotella (Fig. 6C). In the ileum mucosa, in anion mode, 3-amino-5-methylhexanoic acid and ellipticine were significantly negatively correlated with the genus unidentified_Enterobacteriaceae, and 4-methylcatechol, trepibutone, and ellipticine were significantly positively correlated with the phylum Firmicutes. In the cation mode, (+)-castanospermine and (Z)-N-[(2R)-2-amino-1-hydroxy-3-{[(1E)-N-hydroxy-7-(methylsulfanyl)heptanimidoyl]sulfanyl}propylidene]glycine were positively correlated with the phylum Firmicutes, and 2-oxohex-4-enoic acid, (Z)-N-[(2R)-2-amino-1-hydroxy-3-{[(1E)-N-hydroxy-7-(methylsulfanyl)heptanimidoyl]sulfanyl}propylidene]glycine, and N-desmethyltramadol were significantly positively correlated with the family Prevotellaceae (Fig. 6D).
Global overview of proteomics
To identify differentially expressed proteins under specific conditions. Within the SADS-CoV P7 infection group, when compared to the control group (A vs. C), we identified 244 differentially expressed proteins, 137 of which were markedly upregulated and 107 of which were significantly downregulated. Notably, the most substantially downregulated proteins in the A vs. C comparison included the 39S ribosomal protein L53 (mitochondrial) and the S100 calcium-binding protein A9 isoform X1.
Analysis of the SADS-CoV P83 infection group versus the control group (B vs. C) revealed 51 differentially expressed proteins, among which 8 were significantly upregulated and 43 were significantly downregulated. The most notably upregulated and downregulated proteins were membrane-associated phosphatidylinositol transfer protein 1 and putative acyl-coenzyme A thioesterase 6, respectively.
Comparing the SADS-CoV P7 infection group with the SADS-CoV P83 infection group (A vs. B), there were 175 differentially expressed proteins, including 141 significantly upregulated and 34 significantly downregulated proteins. The adenylate cyclase type 5 isoform X1 was the most significantly upregulated protein, whereas the pleckstrin homology-like domain family A member 2 was the most significantly downregulated.
Enrichment analysis utilizing the KEGG pathway database was conducted on differentially expressed proteins to elucidate their involvement in specific biological pathways. For proteins differing between the SADS-CoV P7 group and the control group, the analysis highlighted pathways such as drug metabolism-cytochrome P450, extracellular matrix (ECM)-receptor interaction, focal adhesion, and cell adhesion molecules (CAMs) as significantly affected (Fig. 7A). In contrast, proteins that differed between the SADS-CoV P83 group and the control group were predominantly associated with pathways such as viral myocarditis and focal adhesion (Fig. 7B). Moreover, the differentially expressed proteins identified when comparing the SADS-CoV P7 group and the SADS-CoV P83 group were largely involved in pathways related to dilated cardiomyopathy, hypertrophic cardiomyopathy, ECM-receptor interaction, focal adhesion, and Staphylococcus aureus infection, revealing the diverse metabolic and pathophysiological processes influenced by different infection scenarios (Fig. 7C).
Analysis of the correlations between the microbiota and proteins
We analyzed the correlation between proteins and bacteria. Protein information is shown in Supplementary Table 2. In the P set, in feces, the genus Alloprevotella was significantly negatively correlated with transmembrane 4 L6 family member 1 and transgelin isoform X1 and significantly positively correlated with adenylate cyclase type 5 isoform X1 and aldo-keto reductase family 1, member C-like 1. Additionally, the genus unidentified_Enterobacteriaceae was significantly positively correlated with N-acetyllactosaminide alpha-1,3-galactosyltransferase isoform 1 and significantly negatively correlated with carbonic anhydrase 2 and tubulin-specific chaperone cofactor E-like protein isoform X6. The genus Fusobacterium was significantly positively correlated with bcl-2-like protein 15, UPF0184 protein C9orf16 homolog, histone H1.0, and significantly negatively correlated with cytochrome P450 2C42 precursor, cytochrome b reductase 1, and cytochrome P450 3A29 (Fig. 8A). In the ileum mucosa, the genus unidentified_Prevotellaceae was significantly negatively correlated with myristoylated alanine-rich C-kinase substrate and significantly positively correlated with angiotensin-converting enzyme 2 isoform X1, glycerol-3-phosphate dehydrogenase [NAD(+)], and cytoplasmic lactase-phlorizin hydrolase. The genus unidentified_Enterobacteriaceae was significantly positively correlated with C-type lectin domain family 14 member A, tropomyosin alpha-1 chain isoform X3, and somatostatin precursor and significantly negatively correlated with phospholipase D4 isoform X1, phytanoyl-CoA dioxygenase, and peroxisomal isoform X1 (Fig. 8B). In the I-V set, in feces, the genus Alloprevotella was significantly positively correlated with adenylate cyclase type 5 isoform X1 and negatively correlated with protein S100-A2. The genus Fusobacterium was significantly positively correlated with XP_001929591.1 and negatively correlated with adenylate cyclase type 5 isoform X1 and NAD(P)H dehydrogenase [quinone] 1. The genus Peptostreptococcus was significantly negatively correlated with the protein S100-A2 (Fig. 8C). In the ileum mucosa, the genus unidentified_Enterobacteriaceae was significantly positively correlated with somatostatin precursors and negatively correlated with integrin alpha-IIb precursors (Fig. 8D).
Correlation networks
From the results, it can be observed that, in the P set, in the feces, TUBB2B was significantly negatively correlated with the phylum Tenericutes, and the phylum Tenericutes was negatively correlated with Com_804_pos (ethamivan) (Fig. 9A). The class Mollicutes was positively correlated with Com_804_pos and ADCY5 (Fig. 9A). In the ileum mucosa, the species Escherichia coli was negatively correlated with 14(S)-HDHA and PECAM1. The familyn Ruminococcaceae was positively correlated with Com_565_pos (N,N-dimethylaniline) and negatively correlated with PECAM1 (Fig. 9B). In the I-V set, no mutually correlated network among microbiota, metabolites, and proteins were identified (Fig. 9C and 9D).
The promotion of E. coli adhesion by SADS-CoV
Analysis of the 16S rRNA sequencing data revealed that the abundance of E. coli was significantly greater in the SADS-CoV P7 group than in the SADS-CoV P83 group and the control group (Fig. 10A). Moreover, proteomic analysis of intestinal tissues from piglets in the SADS-CoV P7 challenge group, SADS-CoV P83 challenge group, and control group revealed that, compared with those in the control group, the differentially expressed proteins in the SADS-CoV P7 challenge group were associated with ECM-receptor interactions (Fig. 10A). Compared with those in the control group, the differentially expressed proteins in the SADS-CoV 83 challenge group were also associated with the ECM-receptor interaction pathway. Compared with control group, proteins related to the ECM pathway, including tenascin (TN), integrin alpha5 (ITGA5), and integrin beta-1 (ITGB1), were significantly upregulated after SADS-CoV infection (Fig. 10A).
An interaction model between SADS-CoV infection and E. coli adhesion was established using IPEC-J2 cells, and the association between host ECM-related proteins and E. coli adhesion was investigated. Changes were analyzed in the mRNA levels of ITGB1, ITGA5, and TN in IPEC-J2 cells infected with SADS-CoV at an MOI of 1 at 1 hour and 7 hours postinfection. The results showed that the expression levels of ITGB1, ITGA5, and TN were significantly upregulated at both 1 hour and 7 hours after infection (Fig. 10A).
In the experiment on the effect of SADS-CoV on Escherichia coli adhesion, using the Escherichia coli fluorescence quantitative counting method established by our laboratory, the gene copy number can be used as the adhesion count of the bacteria. Simultaneous inoculation of both virus and E. coli serotype O157 resulted in a significant increase in adhesion counts at 1 hour and 7 hours compared to inoculation with E. coli O157 alone (p < 0.05) (Fig. 10B). Using fluorescence quantitative PCR, the transcription levels of ITGB1 and ITGA5 were detected in IPEC-J2 cells that were either separately inoculated with SADS-CoV or E. coli O157 or simultaneously inoculated with SADS-CoV and E. coli O157. At 1 hour postinfection, there was no significant difference in the expression level of ITGB1 between the coinfection group and the group inoculated solely with the virus (virus-only group) (Fig. 10C). However, the expression level of ITGB1 in the coinfection group was significantly upregulated compared to the level in the group inoculated solely with bacteria (bacteria-only group) (p < 0.001); ITGB1 expression was also significantly upregulated in the virus-only group when compared to expression in the bacteria-only group (p < 0.001) (Fig. 10C). At 7 hours postinfection, there was no significant difference in ITGB1 expression between the coinfection group and the virus-only group. However, the expression level of ITGB1 in both the coinfection group and the virus-only group remained significantly upregulated compared to the level in the bacterial-only group (p < 0.001) (Fig. 10C). At 1 hour postinfection, there was no significant difference in the expression level of ITGA5 between the coinfection group and the bacteria-only group. However, compared to the level in virus-only group, the expression level of ITGA5 was significantly downregulated in the coinfection group (p < 0.05), and the expression of ITGA5 was significantly lower in the bacteria-only group compared to that in the virus-only group (p < 0.01). At 7 hours postinfection, the expression level of ITGA5 was a significantly lower in the bacteria-only group compared to that in the coinfection group. However, compared to the virus-only group, there was a significantly upregulated in ITGA5 expression in the coinfection group (p < 0.005), and the expression level of ITGA5 was significantly downregulated in the bacteria-only group compared to the virus-only group (p < 0.001). Additionally, coinfection led to upregulation of ITGA5 expression when compared to the level in the bacteria-only group (Fig. 10C). These results demonstrated that, at the transcriptional level, the expression patterns of ITGB1 and ITGA5 were largely consistent with the observed increase in bacterial adhesion.
IPEC-J2 cells were subjected to overexpression of ITGA5, followed by bacterial inoculation for adhesion quantification and transcription level measurement. Compared to levels in the control group, ITGA5 overexpression resulted in a significant increase in bacterial adhesion at 1 hour postbacterial inoculation (p < 0.05), along with a highly significant increase in ITGA5 transcriptional levels (p < 0.001) (Fig. 10D). At 7 hours postbacterial inoculation, in the group with ITGA5 overexpression, there was a significant increase in bacterial adhesion compared to in the control group (p < 0.05), accompanied by a highly significant increase in ITGA5 transcriptional levels (p < 0.001) (Fig. 10D). These results indicate that increased expression of ITGA5 leads to increased adhesion of E. coli O157 to cells. At 1 hour after bacterial inoculation, there was no significant difference in bacterial adhesion between the ITGA5-silenced group and the control group, while the transcriptional level of ITGA5 decreased significantly (p < 0.01) (Fig. 10E). At 7 hours postbacterial inoculation, in the ITGA5-silenced group, bacterial adhesion significantly decreased compared to that in the control group (p < 0.05), while there was no significant difference in the transcriptional level of ITGA5 (Fig. 10E). Based on these results, we can conclude that silencing ITGA5 leads to a decrease in bacterial adhesion. Our combined results from the overexpression and silencing of ITGA5 suggest that this gene is a key factor in secondary bacterial infection following SADS-CoV infection in piglets and that increased expression of ITGA5 leads to an increase in E. coli adhesion.
{kind=link}
{kind=link}
{kind=link}
{kind=link}