2.1 Structure‑based strategies applied for screening PDE4 inhibitors
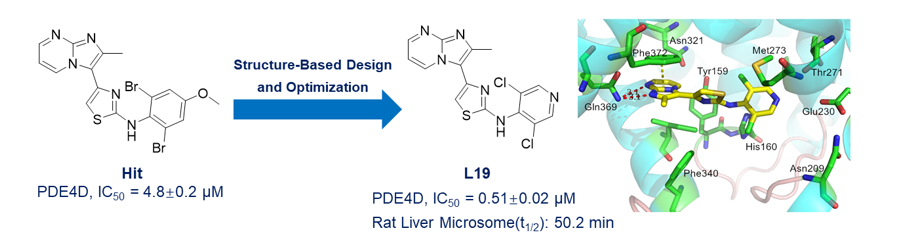
In order to screen novel PDE4 inhibitors, a dataset of 2,530 PDE4 inhibitors was collected from the literature report. By using the Partial Least Squares (PLS) algorithm, we developed a molecular predictive model for the activity prediction of anti-PDE4. As shown in Fig. 1A, the optimal predictive model achieved R² values of 0.812 for the training set and 0.731 for the test set, suggesting robust predictive performance. We then applied this model to predict the activity of 50 compounds procured by our laboratory, finding that the hit compound PTC-209, N-(2,6-dibromo-4-methoxyphenyl)-4-(2-methylimidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amine (Fig. 1B), had the highest predicted pIC50 of 9.38. PTC-209 was initially developed as a selective anti-tumor agent mediated by Bmi-1 [11, 12]. To our knowledge, this is the first report of PTC-209 exhibiting PDE4 inhibitory activity. Subsequent bioassays confirmed that PTC-209 exhibited considerable inhibitory affinity against PDE4, with an IC50 of 4.8 ± 0.2 µM.
2.2 Chemistry
In order to understand its structure–activity relationship (SAR) around the novel scaffold and improve the inhibitory activities, four series of 4-(imidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amine derivatives were designed and synthesized (Schemes 1–4). As shown in Scheme 1, -(2-methylimidazo[1,2-a]pyrimidin-3-yl)ethan-1-one (3) was synthesized by the reaction of pyrimidin-2-amine (1), 1,1-dimethoxy-N,N-dimethylethylamine (2), and chloracetone in toluene [13]. Then, compound 3 was brominated with Br2 in acetic acid [14] to afford the α-bromo ketones (4) and followed by the cyclization with substituted thioureas to get the product L1-L3 [15], respectively. Meanwhile, the α-bromo ketones 4 reacted with the N-Boc thiourea to get the N-Boc-4-(imidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amine (5). And the Boc group of 5 was deprotected by TFA in tetrahydrofuran to afford 4-(imidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amine (L4) [16], which was treated with acetic anhydride in pyridine or treated with carboxylic acids and HATU in the presence of DIPEA to obtain the product L5-L9 [17–19], respectively.
Finally, the same procedures as Schemes 1–2 were used to yield L10-L22, which was firstly synthesized the N-aryl thioureas by the reaction of amines and ammonium thiocyanate in acetic acid [20] and followed by the cyclization with 4 to get the product L10-L22 [21, 22], respectively (Schemes 3–4).
Reagents and conditions: (a) (i) PhMe, 100℃, 3 h; (ii) Chloracetone, rt, 12 h; (b) (i) AcOH, Br2, 110℃, 3 h; (ii) rt, 12 h; (c) Thioureas, EtOH, reflux, 12 h.
Reagents and conditions: (a) N-Boc thiourea, EtOH, Et3N, reflux, 12 h; (b) TFA, THF, rt, 1 h; (c) Ac2O, pyridine, 100℃, 1 h; or RCOOH, HATU, DIPEA, DMF, 100℃, MW, 10 min.
Reagents and conditions: (a) NH4SCN, AcOH, 73℃, 12 h; (b) 5, EtOH, reflux, 12 h.
Reagents and conditions: (a) NH4SCN, AcOH, 73℃, 12 h; (b) 5, EtOH, reflux, 12 h.
2.3 Structure–activity relationships (SARs)
Herein, to understand the structure–activity relationships (SARs) and improve the potency around the 4-(imidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amine scaffold, 22 derivatives were synthesized. Their inhibitory activities against PDE4 are shown in Table 1 and the inhibitory curves of compounds 1, L19, L20, and Rolipram against the PDE4D are shown in Fig. 2, protocols for the expression and enzyme assay of PDE isoforms were similar to our previous works [23–25]. And for the measurement of IC50, eight concentrations at least were used and IC50 values were calculated by the nonlinear regression. In this study, our initial investigations of the SARs of 4-(imidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amines were carried out by substituting the amino position with alkyl or acyl group. However, all the compounds (L1-L9) exhibited weaker PDE4 inhibitory activities than compound 1, indicating that alkyl or acyl substitution at this position is unfavorable for the formation interactions and this subpocket is more favorable for holding an aromatic ring. Subsequently, we focus on change the aryl groups of the amino position. As a result, most of the derivatives (L10-L22) show considerable inhibitory potencies. Specifically, compounds L19 and L20 have a different substitution, in which the introduction of 3,5-dichloropyridin-4-yl/3,5-difluoropyridin-4-yl groups (L19 and L20, IC50 = 0.51 ± 0.02 and 2.1 ± 0.2 µM, respectively) resulted in tighter binding than that with other groups. Finally, compound L19 bearing a 3,5-difluoropyridin-4-yl group has the best inhibitory activity with an IC50 of 0.51 ± 0.02 nM, suggesting that a 3,5-difluoropyridin-4-yl group at this position was favorable for the formation of interactions in the binding pocket.
Table 1. Inhibitory Activities of 4-(imidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amines towards PDE4.
2.5 Rat Liver Microsomal Stability
Metabolic stability is an important parameter to be optimized during the process of design and synthesis of new active compounds [26], and the metabolic stability in human/rat liver microsomes are extensively used in the pharmaceutical industry. Herein, we examined the 4-(imidazo[1,2-a]pyrimidin-3-yl)thiazol-2-amines (1, L11, and L17-L19) using a standard microsomal stability assay with comparison to the testosterone control compound. The results shows that all tested compounds have moderate metabolic stability except L11. Among them, L19 has the best metabolic stability with a t1/2 of 50.2 min and metabolic bioavailability (MF) of 52.7%, which is significantly better than those for the positive control (t1/2 of 2.5 min, and MF of 5.3%).
Table 2
Metabolic Stability of Compounds in Rat Liver Microsomes.
|
Compounds
|
k
|
t1/2 (min)
|
CLint (mL·min− 1·mg− 1)
|
CLh (mL·min− 1·kg− 1)
|
MF (%)
|
|
testosteronea
|
0.2765
|
2.5
|
995.4
|
52.3
|
5.3
|
|
1
|
0.0272
|
25.5
|
97.9
|
35.3
|
36.1
|
|
L11
|
0.0529
|
13.1
|
190.4
|
42.8
|
22.5
|
|
L17
|
0.0357
|
19.4
|
128.5
|
38.6
|
30.1
|
|
L18
|
0.0337
|
20.6
|
121.3
|
37.9
|
31.3
|
|
L19
|
0.0138
|
50.2
|
49.7
|
26.1
|
52.7
|
aTestosterone was the positive control; CLint: intrinsic clearance; CLh: hepatic clearance; MF: metabolic bioavailability.
{kind=link}