Despite recent studies that have further discovered the microbiome in different phenotypic subgroups of CRS, there is still a lack of in-depth research on how the microbiome in the nasal cavity changes during AECRS. A recent study by Vandelaar et al. pointed out that no microbiology differences were noted during AE among the different CRS phenotypes (CRS with nasal polyps, CRS without nasal polyps, and allergic fungal rhinosinusitis)24; meanwhile, Liang et al. found that nasal microbiome significantly altered in eosinophilic CRS as compared to non-eosinophilic CRS28. These results suggested that the inflammation endotype rather than the clinical phenotype may be associated with the nasal microbiome. Due to the insufficient understanding of the microbiology of AE and a lack of objective diagnostic criteria, a short-term antibiotic was usually recommended only based on physician experience, which may promote antibiotic resistance, biofilm formation, and disruption of the natural microbiota29. Thus, exploring the patterns of nasal dysbiosis during AE via 16S rRNA sequencing is necessary, which might promote future precision treatment.
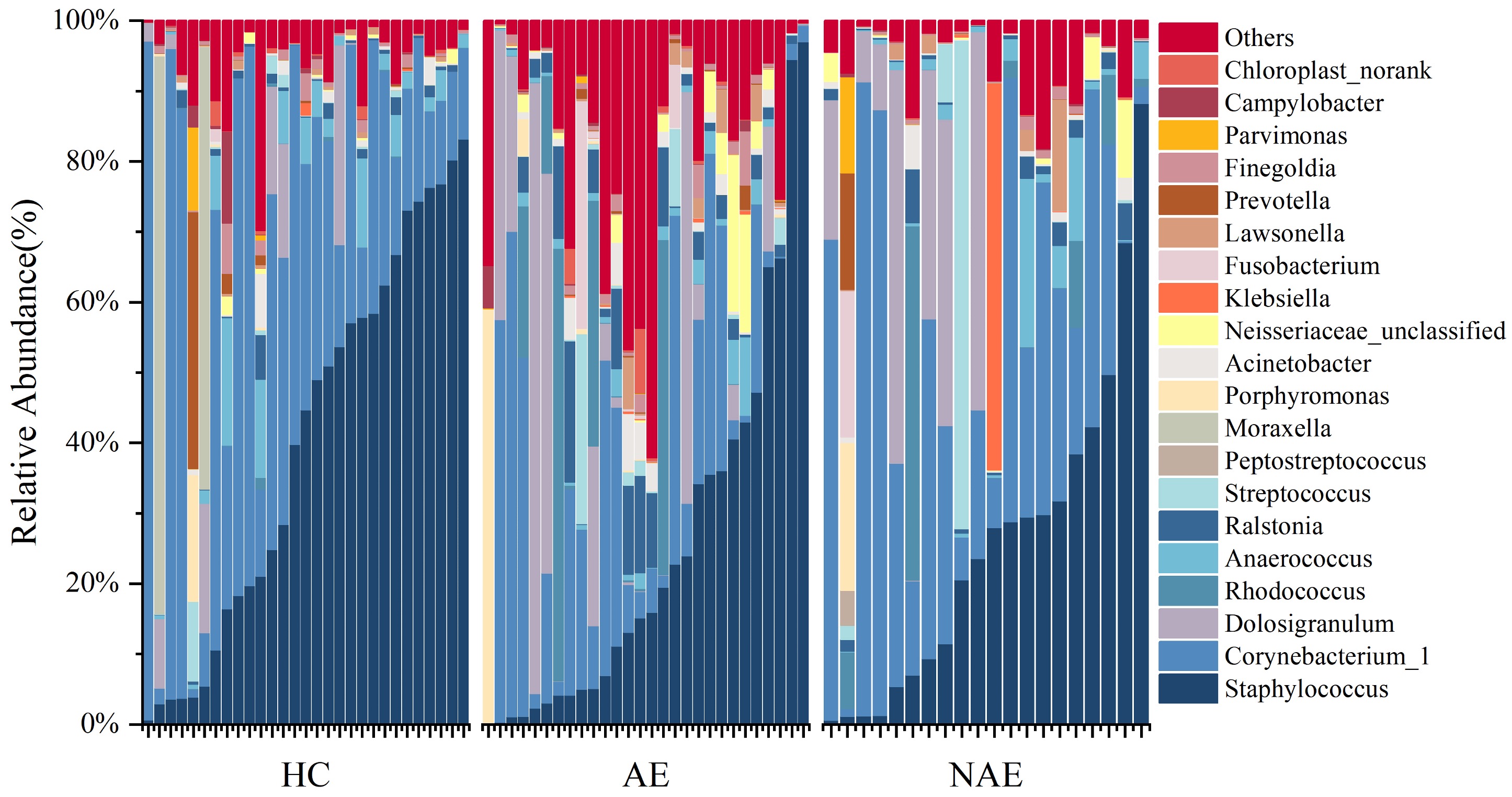
Here, we first identified significant differences in microbiome composition among AECRS, healthy controls, and CRS without AE at the phylum level. The differences were mainly found in phyla Proteobacteria and Bacteroidetes. At the genus level, our finding was consistent with previous findings6, 29. Although Staphylococcus and Corynebacterium_1 remained the core composition in nasal microbiome among the three groups, we found that the abundance of several important pathogenic bacteria in AECRS varied significantly, including the Ralstonia and Acinetobacter. These genera did alter remarkably according to LEfSe analysis, which had been proved to be related to the development of CRS8,9,16,22. Corynebacterium and Staphylococcus were recognized as core sinus microbiome in CRS5. Evidence for an inverse relationship between Staphylococcus aureus and Corynebacterium supported that Corynebacterium protected patients from respiratory illness and exacerbation30–33. Previous studies found that Staphylococcus aureus was one of the predominant bacteria in the sinus microbiome in AECRS patients8, 15, 29. Therefore, the decreased Corynebacterium in AE may increase the risk for AE by provoking Staphylococcus aureus to switch from symbiotic to toxic, which needs further confirmation by more advanced sequencing methods and interpretation at the species level. The loss of dominant bacteria, especially Corynebacterium, laid the foundation for other pathogenic bacteria’s invasion and colonization and finally led to dysbiosis34.
Association between inflammation parameters, disease, and severity measurements were also analyzed. Interestingly, genera with MRA > 0.1% barely correlated with disease activity and severity. However, the disease severity measurements have closer relations to rare constituents (MRA < 1%), indicating that the heterogeneity and the development of AECRS may depend more on rare constituents. Facklamia was isolated from milk and cow samples and reported in mattress dust to be relatively abundant in farming environment35. Xanthomonas has been reported in AECRS8 and was associated with impaired respiratory function in pulmonary emphysema patients36. Legionella was a typical pathogen for the AE of different airway disease37, 38. Our study found that Xanthomonas and Legionella were positively related to eosinophil count and EDN level, indicating that they may play an essential role in developing eosinophil inflammation. Evidence has shown that bronchial hyperresponsiveness was associated with Sphingomonadaceae39 due to the activation of iNKT cells by glycosphingolipids. Faecalibacterium was reported to be dominant in allergic rhinitis40 and was enriched in children with severe asthma41. For the first time, our study proved that the above microbiota of rare constituents in the nasal cavity was correlated with disease severity among patients with AECRS. Considering that an increased Shannon index was found in AECRS and the different genera are mostly rare compositions with MRA < 1%, we further calculated the OTU amount among the three groups, and the results showed no difference, indicating there may be more related to the variation in microbiome composition.
Our research used PICUST to predict the variations in the metabolism pathways of dysbiosis. In previous research, bacterial-associated LPS was found to induce the expression of oncostatin M in severe asthma, leading to airway inflammation and mucus hypersecretion42. And LPS regulated the eosinophilic airway inflammation via the COX-2/PGE (2) axis in the mechanism of CRS43. Therefore, our finding of enhanced LPS biosynthesis during AE compared to CRS without AE and healthy controls proved that the LPS was involved in the pathology of inflammation in AE. Previous studies reported that cysteine oxidations contributed to chronic inflammation and the development of asthma, and the level of cysteine significantly decreased in plasma of children with severe asthma44, 45. Methionine is a critical amino acid required for polyamine biosynthesis, essential for fast-growing bacteria, which must constantly synthesize polyamines to replicate their DNA46. Our study found that cysteine and methionine metabolism decreased significantly in patients with AECRS compared to healthy controls and CRS without AE. Fatty acid metabolism and glycolytic pathways were previously found to play essential roles in asthma pathology47. Further studies are required to explore the molecular mechanism and extent of action of the above metabolism pathways in developing AECRS.
Current diagnosis criteria and indications for treatment for AECRS were based on the sudden worsening symptoms9. Our research confirmed the effectiveness of typical clinical tools in assessing the disease severity of AECRS, such as scales for symptoms, nasal endoscopy, and CT scans. Besides, our research found that EDN increased significantly during AE and presented a significantly positive association with disease severity. The ROC curve with the AUC value of 0.820 suggested that EDN may serve as a biomarker for AECRS. As the dysbiosis of nasal microbiota played a necessary role in AE, utilizing nasal microbial signatures to discriminate CRS disease status has great potential. The prediction model based on microbiota for disease state and diagnosis has been previously explored in the recurrence of CRSwNP48. In our study, a model comprising of Klebsiella, Xanthomonas, Achromobacter, Sphingobium, Legionella, Cellvibrio, Pseudomonas, Flaviflexus, Enterococcus, Thermobrachium, Atopostipes, Vagococcus, Neochlamydia and Fervidobacterium was able to diagnose AE from CRS without AE and healthy controls. These findings suggested the possibility of noninvasive nasal sample profiles to diagnose AE among patients with CRS.
The innovations of our study included first utilizing 16S rRNA to identify the microbiome dysbiosis of AECRS, regardless of phenotypes, and aiming to find a universe variation during AE. Nevertheless, this study has several limitations. First, the sample size was relatively small and this was a single-center study. A multicenter study with large sample size will be needed. Second, further validation cohorts are needed to confirm that the nasal microbiome-based classifier can accurately distinguish patients with AECRS and can identify patients with different inflammation status.
{kind=link}