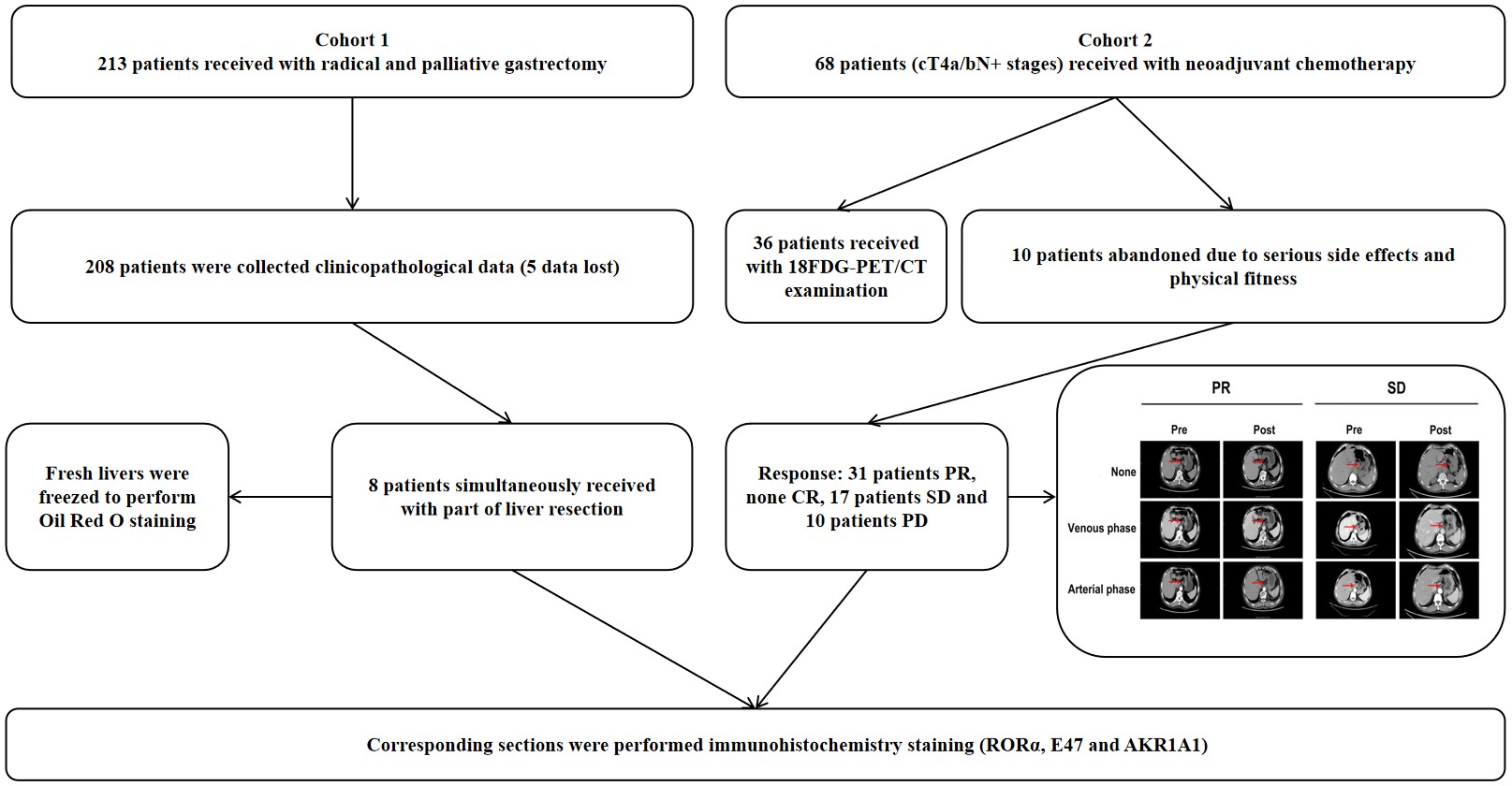
Patients and specimens
Samples (paraffin-embedded sections and frozen liver tissues) and clinicopathological data of GC patients were collected from The First Affiliated Hospital of Anhui Medical University (Hefei, Anhui) from August 2016 to September 2023. The inclusion criteria for eligible patients were confirmed gastric adenocarcinoma and Eastern Cooperative Oncology Group Performance Status scores between 0 and 2 (15). The clinical stage was assessed by chest, abdomen and pelvis enhanced CT or MRI according to the 7th Edition of the International Union against Cancer Tumor–Node–Metastasis (TNM) classification(16). The standard uptake value (SUV) was determined by 18F-FDG PEC/CT. Imaging diagnosis was determined by at least two radiologists from the Department of Nuclear Medicine. The collection diagram and demographic characteristics are illustrated in Figure S1 and Table 1. Disease-free survival (DFS) time was defined as the time of local recurrence, distant metastasis or death from any complication. The response to treatment was defined as partial response (PR), complete response (CR), stable disease (SD) or progressive disease (PD) in locally advanced GC patients (cT4a/bN+) who received neoadjuvant chemotherapy according to the RECIST 1.1 guidelines (17). All patients signed informed consent before the study, which was approved by the human Ethics Committee of Anhui Medical University (20180344, Hefei, Anhui).
Neoadjuvant chemotherapy regimens
Patients received 3 cycles of treatment based on the XELOX and FOLFOX-6 regimens according to the Chinese Society of Clinical Oncology (CSCO) and National Comprehensive Cancer Network (NCCN) guidelines as follows (18,19). 1): The XELOX regimen included oxaliplatin (120 mg/m2 on day 1 in each cycle, intravenously) plus capecitabine (1000 mg/m2 twice daily on days 1-14, orally); 2): The FOLFOX-6 regimen included fluorouracil (400 mg/m2 on day 1 followed by 2400 mg/m2 on46 hoursin each cycle, intravenously), leucovorin (400 mg/m2 on day 1 in each cycle, intravenously) plus oxaliplatin (400 mg/m2 on day 1 in each cycle, intravenously).
Single-cell sequencing data downloading, preprocessing and analysis
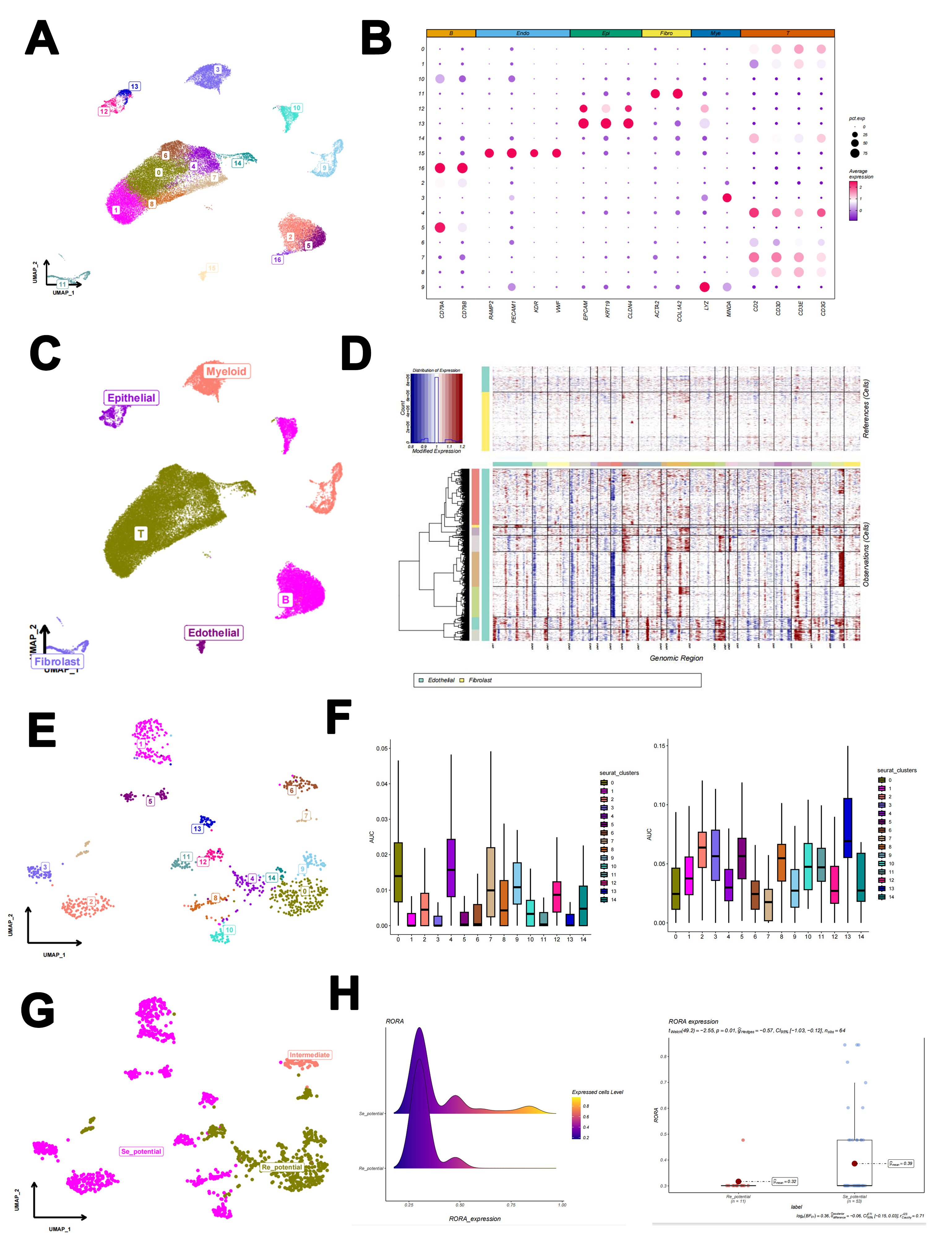
Single-cell RNA transcription data were downloaded from the NCBI Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo/) with 10X Genomics in 10 fresh tissues (GSE163558), including 3 primary tumor tissues, 1 adjacent gastric tissue and 6 tissues with GC cell metastasis (2 lymph nodes, 1 ovary, 1 peritoneum and 2 livers). These tissues were invaded by GC cells. The functions of Read10x, CreatSeuratObject and Merge in the R package ‘Seurat’ were utilized to create data for R software to obtain high-quality cells.
DoubletFinder was used to remove double cells. The inclusion criteria were as follows: fewer than 80000 total genes, fewer than 5% mitochondrial genes and fewer than 50 housekeeping genes.
The top 2000 highly expressed genes were identified through the function FindALLMarkers according to dispersion and average expression levels. The functions Normalizedata and Harmony were utilized to standardize scaling and eliminate batch effects, respectively. The functions FindNeighbors and FindClusters were utilized to gather different clusters through differentially expressed genes at a resolution of 0.6.
The copy number analysis was performed by the R package inferCNV (v1.0.6) using the default parameters. Endothelial and fibroblasts were used as controls to identify highly CNV epithelial cells, which were regarded as cancer cells.
The 123 5-FU-related genes were collected from published references, including 83 resistance genes and 40 sensitive genes (20). The AUCell algorithm was utilized to score each tumor cell line. Based on the scores, we nomenclatured each cluster to compare the expression levels of RORα among these cell types. The ridge and box plots were generated using the ggolot package and ggstatsplot package, respectively.
RNA transcriptome analysis
The RNA-sequencing transcriptome was generated by OE Biotech Co., Ltd. (Shanghai, China) in the primary and adjacent tissues of 5 GC patients. Total RNA was extracted with TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s instructions. RNA purity and quantification were performed using a NanoDrop 2000 Spectrophotometer (Thermo Scientific, USA). RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent, Technologies, Santa Clara, CA, USA). Libraries were generated with a VAHTS Universal V6 RNA-seq Library Prep Kit according to the manufacturer’s instructions.
Transcription factor prediction via bioinformatic analysis
The online bioinformatics tools AnimalTFDBI, TRANSFAC PATCH and MATCH were used to identify the potential transcription factors of AKR1A1 (Table S1). JASPAR (http://jaspar.genereg.net/) was used to evaluate the highest binding score of the two candidates.
Cell culture and treatment
Human GC cell lines (AGS and MKN-74) and a mouse forestomach carcinoma (MFC) cell line were obtained from Procell Life Science and Technology (Wuhan, China). These cells were cultured according to the protocol. Cells were treated with SR1078 (5 µM, GlpBio, Montclair, CA, USA), 2-DG (8 mM, Sigma‒Aldrich, MO, USA) and atorvastatin (10 µM, GlpBio, Montclair, CA, USA) for 24 h. SR1078 was dissolved in phosphate-buffered saline (PBS). Other reagents, including fluorouracil (GC14466, GlpBio, Montclair, CA, USA), were dissolved in dimethyl sulfoxide (DMSO).
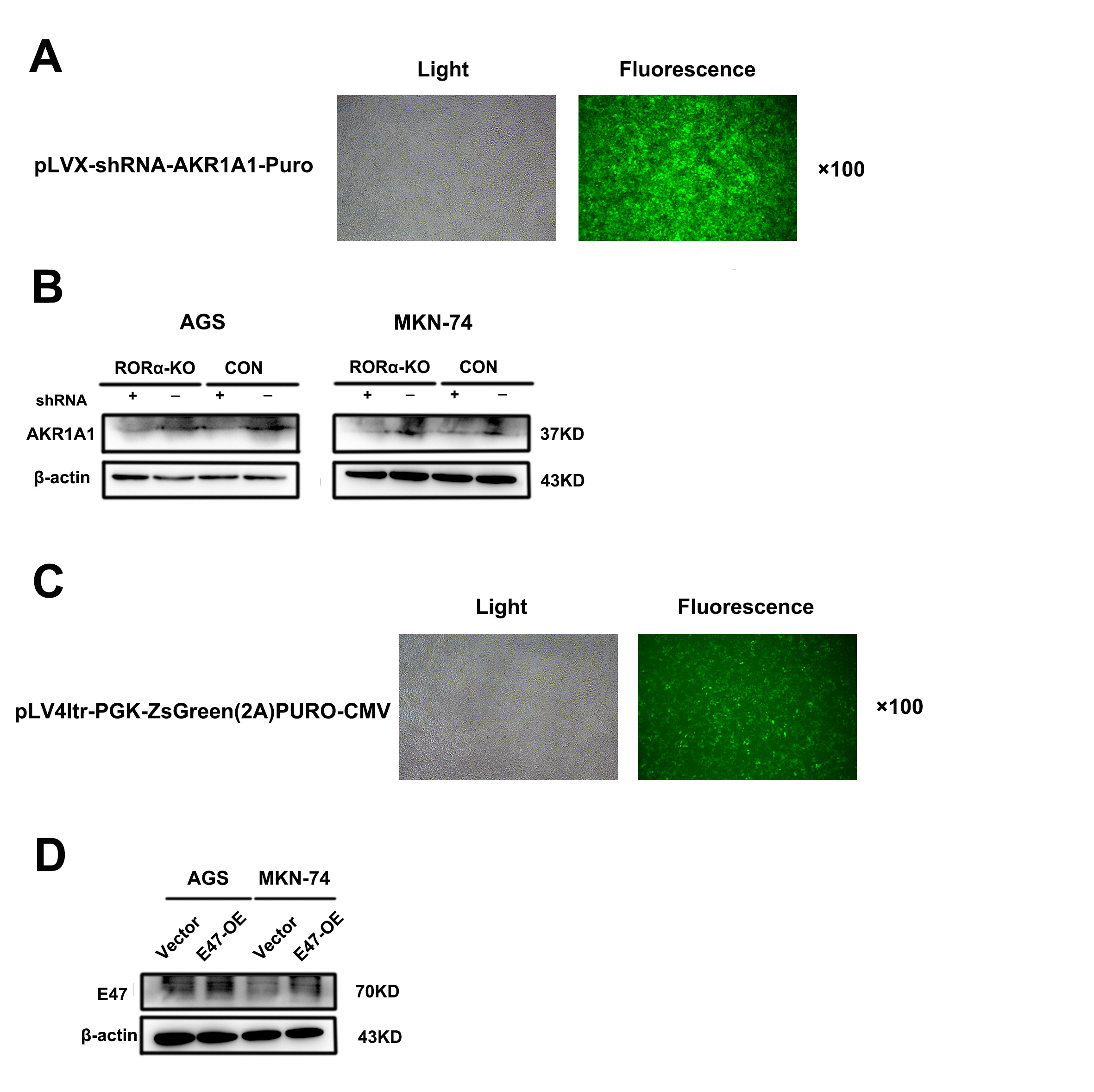
CRISPR-Cas9 gene deletion and small interfering RNA (siRNA) transfection
Single guide RNA (sgRNA) oligonucleotides were cloned and inserted into LV-U6-spsgRNA(RORα)-CMV-SV40-NLS-spCas9-NLS-Flag-P2A-Puro-T2A-EGFP-WPRE (human) and packaged as lentiviruses (BrainVTA, Wuhan, China). An alignment of the DNA sequences of the human and mouse RORα genes revealed 91% similarity according to the NCBI database. The cells were infected with a virus expressing Cas9 and gRNA for 12 hours. After culturing with 2 μg/ml puromycin for 72 h, we found that the fluorescence (30-50%) of the AGS (MOI=0.5), MKN-74 (MOI=0.3) and MFC (MOI=0.5) strains was satisfactory. Western blotting was used to detect protein expression levels after transfection. The RORα-KO sgRNA sequence (human) was GTAATCGACAGTGTTGGCAG. siRNA-E47 (RiboBio, Guangzhou, China) and siRNA-β-catenin (RiboBio, Guangzhou, China) were dissolved in 1,2-dierucoyl-sn-glycero-3-phosphocholine (DEPC). The cells were cultured in 1640 medium without fetal bovine serum (FBS). The siRNAs and Lipofectamine 3000 (Thermo Scientific, USA) were diluted and incubated at room temperature for 30 minutes. The mixtures were cultured for 6 h in an incubator, and then the medium was changed to complete medium for 24 h until the next series of experiments.
Lentivirus infection for knockdown and overexpression
The lentiviral particles pLVX-shRNA-AKR1A1-Puro (LVP0916) and pLV4ltr-PGK-ZsGreen(2A)PURO-CMV (LVP0917, E47 overexpression) were purchased from TSINGKE (Beijing, China). Cells were seeded into 6-well plates and cultured with 8 μg/ml polybrene complete medium for 24 h. Next, AGS (MOI=10, 10) and MKN-74 (MOI=5, 5) cells were transfected with virus for knockdown or overexpression, respectively. The cells were cultured in an incubator for 24 h, after which the medium was changed to complete medium for western blot analysis to determine the transfection efficiency.
Colony formation assay
Cells (1200 cells/well) were seeded into 6-well plates and cultured in an incubator through a counting plate. The wells were fixed and stained with 0.1% crystal violet for 10 min after 10 days. The number of colonies in each well was calculated three times.
Cell Counting Kit-8 (CCK-8) assay
Cells were seeded into 96-well microplates and cultured with fluorouracil according to concentration gradients. The mixtures were incubated in 10 µl of enhanced Cell Counting Kit-8 (CCK-8, C0042, Beyotime, Haimen, China) for 1 h according to the manufacturer's protocol. The optical density (OD) values were measured at 450 nm using a microplate reader (Thermo Scientific, USA). The relative absorbance values were analyzed through OD values.
Flow cytometry analysis
Pretreated cells were cultured with fluorouracil (50 µg/ml) for 24 h. The cell suspension was collected and then incubated with 5 µl of Annexin V-FITC and 5 µl of PI through an Apoptosis Kit (E-CK-A211, Elabscience, Wuhan, China) at room temperature for 15 min. The percentage of cell apoptosis was determined through flow cytometry (Beckman Coulter, USA).
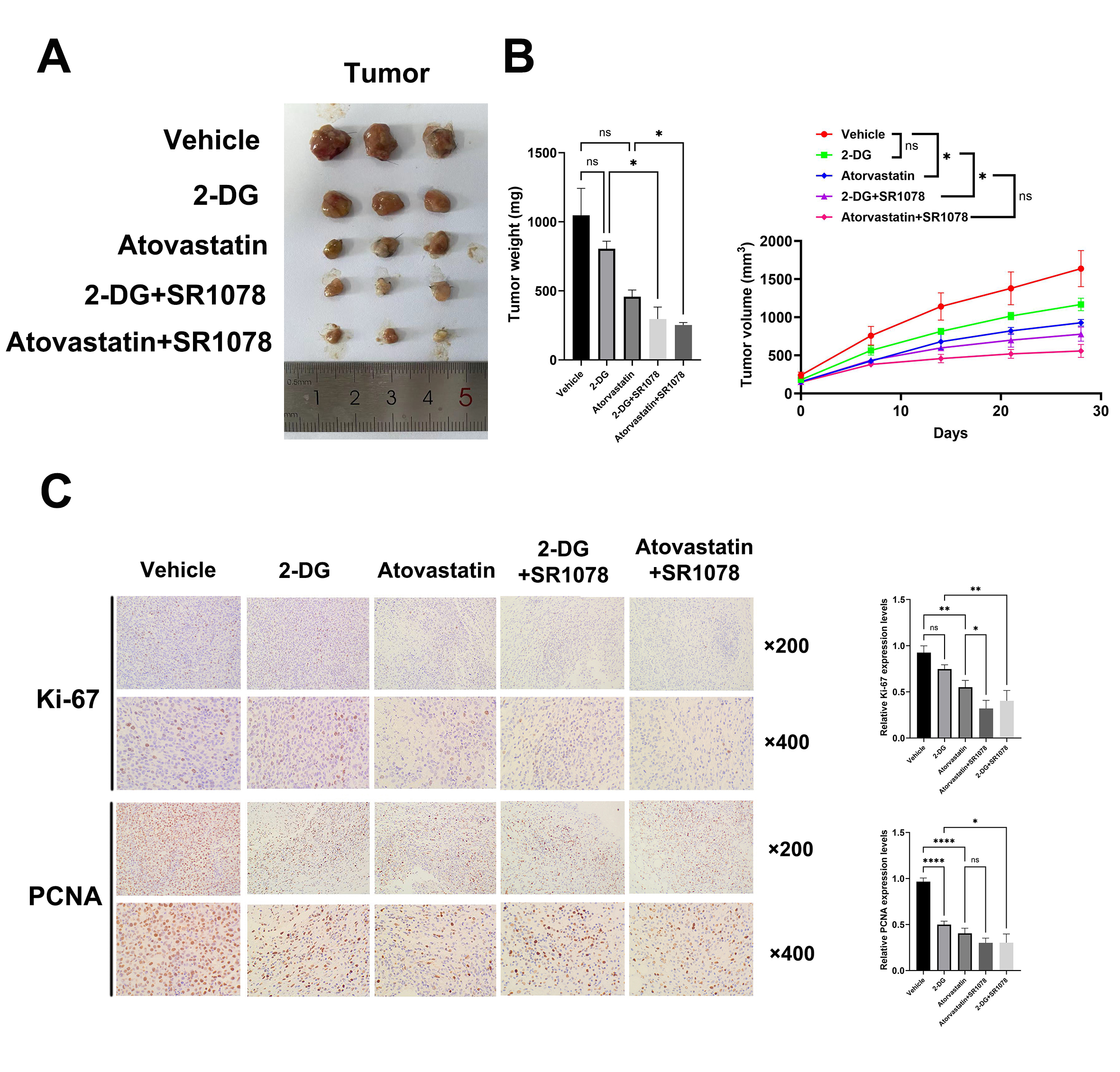
Mouse model construction
Six- to eight-week-old mice (C57, Vital River Laboratory Animal Technology, Beijing, China) were randomly divided into different groups. A total of 1 × 106MFC cells were injected into the subcutaneous flank of mice to construct a subcutaneous tumor model for 4 weeks. A total of 5 × 105 MFC cells were injected into the portal or caudal vein of mice to establish a model of liver metastasis or necrosis for 2 weeks. The weight and volume (1/2 × long diameter × short diameter) of the mice were measured weekly, and the mice were sacrificed, and samples were collected for immunohistochemistry and hematoxylin-eosin (H-E) staining after an adequate breeding time. The liver metastatic nodules and extent of necrosis were observed and calculated by microscopy (OLYMPUS, Japan). These studies complied with international protective guidelines for laboratory animals and ethical standards. This project was approved by the Ethics Committee of Anhui Medical University (20180365, Hefei, Anhui).
Immunohistochemistry, H&E and Oil Red O staining
The sections were deparaffinized and immersed in boiling citrate buffer (pH=6.0). Primary antibodies against RORα, Ki-67, proliferating cell nuclear antigen (PCNA), multidrug resistance 1 (MDR1), E47 and AKR1A1 were incubated with the cells for 2 h (Table S2). 3,3'-diaminobenzidine (ZSGB‑BIO, OriGene Technologies, Beijing, China) and 20% hematoxylin were used for staining at room temperature. The mean optical density (MOD) method was used to assess relative expression levels in previous studies (21). High and low expression was judged as the proportion grade multiplied by the staining intensity score. The proportion grades were as follows: 3 (> 76%), 2 (26–75%), 1 (26–50%), and 0 (< 25%). The staining intensity scores were as follows: 2 (brownish and yellow brown), 1 (light staining) and 0 (no staining). A sample score of 0–2 was defined as low expression, and 3–6 was defined as high expression. Frozen liver tissues were collected, and Oil Red O staining was performed by Servicebio Company (Wuhan, China). A TissueGnostics viewer (TG, Austria) was used to obtain images, which were quantified with Image J software.
Immunofluorescence colocalization
Cells were seeded into 6-well plates containing slides for 24 h. The slides were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.5% Triton X-100 for 10 min and then blocked with bovine serum albumin (BSA) for 2 h at room temperature. After washing with PBS three times. The blocked cells were incubated with primary antibodies against β-catenin and E47 at 4°C overnight and then incubated with fluorescent secondary antibodies for 1 h at room temperature (Table S2). 4′,6-Diamidino-2-phenylindole (DAPI) was used to stain the nuclei for 10 min. TG was used for image acquisition.
Western blot analysis
Cell lysis was performed with RIPA buffer. The protein concentration of the samples was determined by a BCA assay (KeyGen Biotech, Nanjing, China). The protein extract was separated by 12% SDS‒PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. Primary antibodies against RORα, AKR1A1, E47, β-catenin, wnt3a and β-actin were incubated overnight at 4°C (Table S2). The membranes were incubated with secondary antibodies for 2 h at room temperature. The bands were imaged with a chemiluminescence system (Tanon, Shanghai, China).
Quantitative real-time PCR (qPCR)
Total RNA was isolated using TRIzol (Invitrogen, USA) according to the manufacturer’s instructions. cDNA was generated using a cDNA kit (TaKaRa, Shiga, Japan), and PCR was performed using GoTaq® Green Master Mix (Promega, USA) on a 7900 Thermal Cycler (Thermo Fisher Scientific, USA) through denaturation, renaturation and extension of primers (Table S3).Relative mRNA expression levels were calculated through the 2−ΔΔCq method(22). The β-actin gene was used as an internal control for normalization.
Seahorse assay
The extracellular acidification rate (ECAR) was determined with a Seahorse XFe24 analyzer (Seahorse Bioscience, USA) according to the manufacturer’s guidelines. Briefly, the pretreated cells were seeded into 24-well plates after sensor calibration. Next, XF test medium was added to each well, and the plates were incubated for 1 h at room temperature. Subsequently, the ECAR was detected following sequential treatment with glucose (10 mM), oligomycin (1 μM), and 2-DG (100 mM).
ATP production measurement
AnATP assay kit (S0026, Biyotime, Haimen, China) was used to detect ATP consumption according to the manufacturer’s instructions. The cell lysate was centrifuged for 20 min at 40°C to collect the supernatant, which was subsequently incubated with diluted ATP detection reagent for 5 min. The relative unit light (RUL) values were measured by a microplate reader and were converted into ATP concentrations according to a standard curve.
Triglyceride and phospholipid measurements
The cellular contents of triglycerides and phospholipids were measured with a triglyceride assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) and an EnzyChrom™ phospholipid assay kit (BioAssay Systems, CA, USA) according to the manufacturer’s instructions. Briefly, the OD values of the lysed cells were measured with a microplate reader at 500 or 570 nm. The concentration was calculated by a standard curve.
Coimmunoprecipitation (Co-IP) assay
The cell supernatant was collected and incubated with antibodies against E47 or β-catenin (Table S2) overnight at 4°C. Protein A/G PLUS-Agarose beads (10μl, sc-2003, Santa Cruz Biotechnology, USA) were added and incubated at room temperature for 1 h. After transient centrifugation. The immunoprecipitated agarose beads were collected and resuspended in loading buffer. Next, these samples were subjected to immunoblotting after boiling.
Chromatin immunoprecipitation (Chip)-qPCR assay
A chip assay kit (P2078, Beyotime, Haimen, China) was used to perform the chip assay. The detailed process of the experiment was described in a previous study(23). The primary antibodies against E47 and β-catenin are listed in Table S2. Immune complexes were obtained after crosslinking, ultrasonication and washing. The purified DNA fragments were obtained through decrosslinking and were subjected to qPCR.
Luciferase reporter assay
The region of the AKR1A1 promoter from +291 to -1664 bp was inserted into the pGL3-basic vector (Addgene, #212936) for construction of the luciferase reporter plasmid. Next, the cells were cotransfected with the E47 overexpression or control vector and luciferase reporter plasmids with Lipofectamine 3000 for 24 h. A luciferase assay was performed with a PierceTM luciferase kit (Promega, USA) according to the manufacturer’s instructions. The sequences used were as follows: GCCAACAGTTACTTTGTTCCCT (forward) and ATGTCCATTTTGATCTGCAAAGT (reverse).
Half-life of E47 mRNA and protein expression
Cells were treated with actinomycin D (5 µg/mL, Aladdin, Shanghai, China) or cycloheximide (20 µg/ml, MCE, USA) for the indicated durations. The mRNA abundance at each time point was normalized to the initial abundance.
Analysis of drug combinations
The effect of drug combination was determined by the SynerFinder 3.0 web application (https://synergyfinder.org/) according to the Zero Interaction Potency (ZIP) model(24). The percentage of relative cell viability was calculated from the OD values. The white and red regions represent superposition and synergy, respectively. The summary synergy scores were as follows: 1) less than -10 indicated antagonistic effects; 2) from -10 to 10 indicated additive effects; and 3) greater than 10 indicated synergistic effects.
Statistical analysis
Statistical analysis was performed with SPSS 19.0 and GraphPad Prism 10.0.0 software. Comparisons between different groups were performed using ANOVA or the Mann‒Whitney test. Survival analysis was performed by the Kaplan‒Meier method and log-rank test. The correlation analysis was performed by the chi-squared test. Univariate and multivariate survival analyses were performed with a Cox proportional hazards model. P value < 0.05 was considered a statistically significant difference.
{kind=link}
{kind=link}
{kind=link}
{kind=link}