Experimental part
Chemistry
Melting points were determined on a Kofler hotstage apparatus and are uncorrected. 1H, 13C and 19F NMR spectra were recorded on a Bruker Avance III HD 400 MHz spectrometer using the indicated deuterated solvents. Chemical shifts (δ) are reported in parts per million and coupling constants (J) are reported in hertz (Hz). 13C NMR spectra were fully decoupled. The following abbreviations were used to explain multiplicities: singlet (s), doublet (d), triplet (t), double doublet (dd), broad (br), and multiplet (m). Chromatographic separations were performed on silica gel columns by flash column chromatography (Kieselgel 40, 0.040−0.063 mm, Merck) or using ISOLUTE Flash Si II cartridges (Biotage). Reactions were followed by thin-layer chromatography (TLC) on Merck aluminum silica gel (60 F254) sheets that were visualized under a UV lamp. Evaporation was performed in vacuo (rotating evaporator). Sodium sulfate was always used as the drying agent. Commercially available chemicals were purchased from Sigma-Aldrich (Merck). The ≥ 95% purity of the tested compounds was confirmed by combustion analysis. Analytical results are within ± 0.4% of the theoretical values. The ESI-MS spectra were recorded by direct injection at 5 (positive) and 7 (negative) μl min−1 flow rate in an Orbitrap high-resolution mass spectrometer (Thermo, San Jose, CA, USA), equipped with HESI source.
Synthetic details and characterization of compounds
General procedure for the condensation reaction using EDC/HOBt system for the synthesis of amides 6, 9, 11, 13, 15
To a solution of the appropriate carboxylic acid (2.25 eq) in dry DCM (5 mL/mmol), the appropriate amine (1 eq), Et3N (2.25 eq), HOBt (2.25) and finally EDC (2.25) were added. The reaction mixture was stirred at room temperature for 12h, under argon atmosphere. Then the solvent was evaporated and EtOAc was added and washed with a saturated solution of NaHCO3, HCl 1N and brine. The organic phase was dried over Na2SO4, filtered and evaporated under reduced pressure.
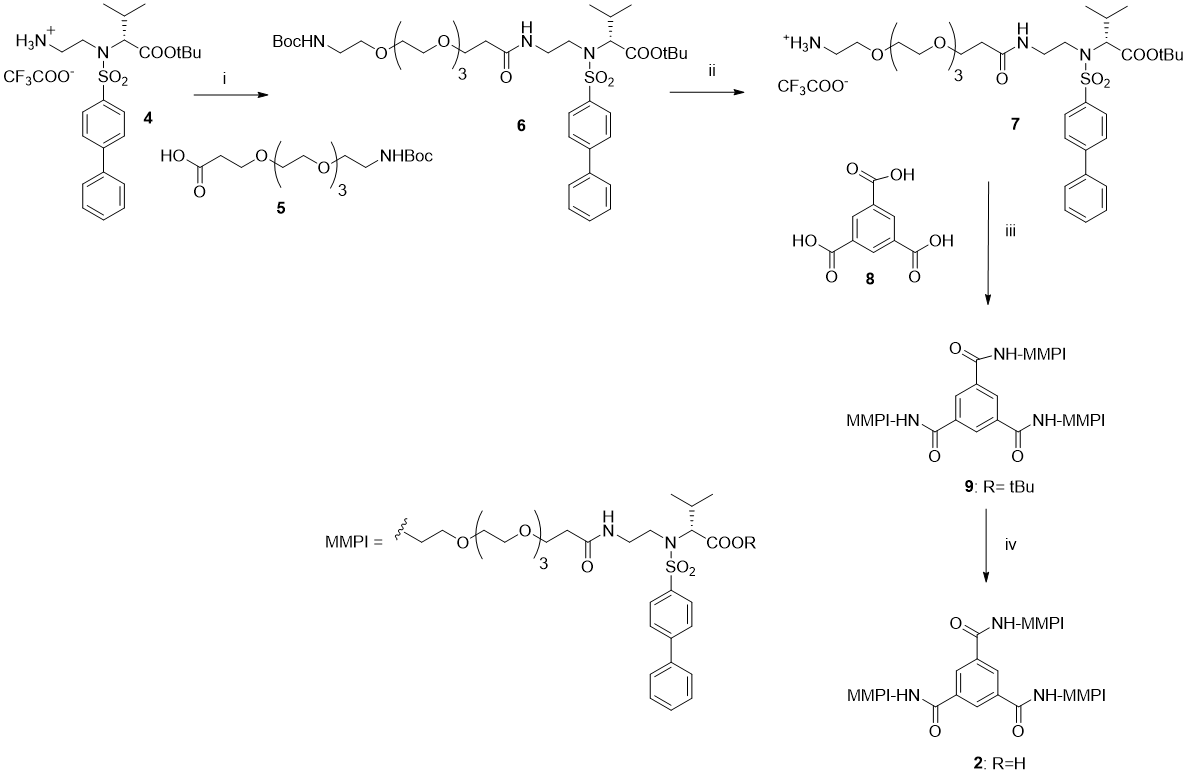
(S)-24-([1,1'-biphenyl]-4-ylsulfonyl)-25-isopropyl-2,2-dimethyl-4,20-dioxo-3,8,11,14,17-pentaoxa-5,21,24-triazahexacosan-26-oic acid (6). The title compound was synthesized from amine 4 and carboxylic acid 5 following the general procedure. The pure compound was obtained as a colorless oil (97% yield). 1H NMR (400 MHz, CDCl3) δ: 7.90-7.88 (m, 2H), 7.69-7.66 (m, 2H), 7.57-7.55 (m, 2H), 7.49-7.41 (m, 3H), 6.99 (bs, 1H), 5.11 (bs, 1H), 4.11 (d, J= 7.2 Hz, 1H), 3.96-3.83 (m, 1H), 3.75 (t, J= 5.6 Hz, 2H), 3.66-3.47 (m, 12H), 3.38-3.29 (m, 4H), 3.19-3.12 (m, 2H), 2.83-2.81 (m, 2H), 2.49 (t, J= 6.0 Hz, 2H), 2.05-2.03 (m, 1H), 1.43 (s, 9H), 1.23 (s, 9H), 1.05, 0.95 (2xd, J= 6.4 Hz, each 3H).
Trimeric derivative 9: The title compound was prepared from amine 7 (130 mg, 0.16 mmol, 4 eq) and the commercial benzene-1,3,5-tricarboxylic acid 8 (8.6 mg, 0.041 mmol, 1 eq) in dry DMF (5mL). Et3N (0.15 mL, 1.11 mmol, 27 eq), HOBt (55 mg, 0.41 mmol, 10 eq), EDC (78.50 mg, 0.41 mmol, 10 eq) were added following the general procedure. The crude residue was purified by reverse phase flash chromatography using a C-18 Isolute cartridge (SI -C18 5g) (H2O /MeOH 1:2) affording the pure compound 9 as a colorless oil (47 mg, 52% yield). 1H NMR (400 MHz, CDCl3) δ: 8.54 (s, 3H), 8.05 (bs, 3H), 7.90-7.87 (m, 6H), 7.68-7.66 (m, 6H), 7.56-7.54 (m, 6H), 7.47-7.40 (m, 9H), 7.11 (bt, 3H), 3.92 (d, J= 7.2 Hz, 3H), 3.81-3.76 (m, 4H), 3.68-3.47 (m, 57H), 3.36-3.34 (m, 3H), 2.39 (t, J= 6.0 Hz, 6H), 2.16-2.11 (m, 3H), 1.21 (s, 27H), 1.02, 0.93 (2xd, J= 6.4 Hz, each 9H). 13C NMR (100 MHz, CDCl3) δ:171.9, 169.7, 166.6, 145.7, 139.3, 138.2, 135.1, 129.1, 129.9, 128.5, 128.1, 127.7, 127.3, 82.1, 70.4, 70.2, 70.1, 70.0, 69.9, 67.0, 66.6, 44.1, 40.3, 40.0, 36.6, 29.2, 27.7, 20.1, 19.3.
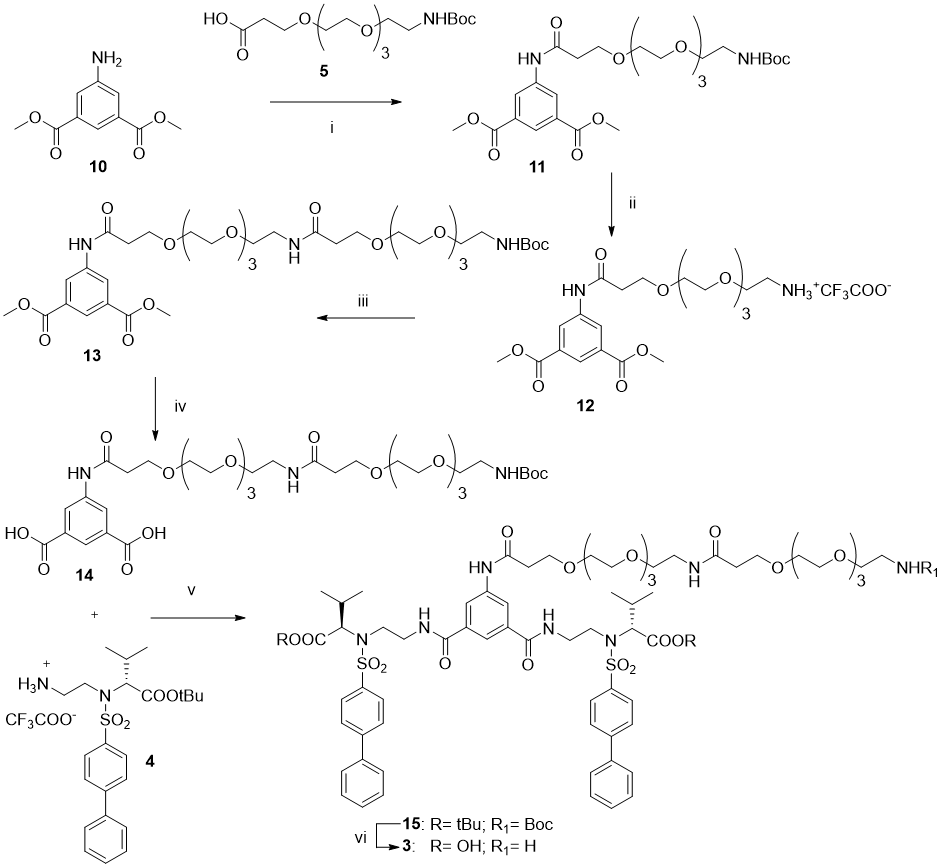
Dimethyl 5-(2,2-dimethyl-4-oxo-3,8,11,14,17-pentaoxa-5-azaicosan-20-amido)isophthalate (11): Amide 11 was synthesized from the commercial Boc-NH-(PEG)3-COOH 5 and dimethyl 5-aminoisophthalate 10 following the general procedure. The crude was purified by flash chromatography using an Isolute cartridge (SI 10g) (100% CHCl3) affording the pure compound as a colorless oil (53% yield). 1H NMR (400 MHz, CDCl3) δ: 9.14 (bs, 1H), 8.44-8.43 (m, 2H), 8.40-8.39 (m, 1H), 3.92 (s, 6H), 3.84 (t, J= 5.12 Hz, 2H), 3.71 (s, 6H), 3.67-3.61 (m, 2H), 3.57-3.55 (m, 4H), 3.52 (t, J= 5.12 Hz, 2H), 3.28 (t, J= 5.12 Hz, 2H) 2.69 (t, J= 5.12 Hz, 2H), 1.43 (s, 9H). 13C NMR (100 MHz, CDCl3) δ: 170.6; 166.2; 156.2; 139.2; 131.3; 126.1; 125.2; 70.8; 70.6; 70.5; 70.4; 70.3; 67.1; 52.5; 40.5; 38.0; 28.6.
dimethyl 5-(2,2-dimethyl-4,20-dioxo-3,8,11,14,17,24,27,30,33-nonaoxa-5,21-diazahexatriacontan-36-amido)isophthalate (13). The title compound was synthesized from amine 12 and Boc-NH-(PEG)3-COOH 5 following the general procedure. The pure compound was obtained as a colorless oil (100% yield). 1H NMR (400 MHz, CDCl3) δ: 9.17 (bs, 1H), 8.46-8.44 (m, 2H, Ar), 8.40-8.39 (m, 1H), 6.81 (bs, 1H), 5.07 (bs, 1H), 3.92 (s, 6H), 3.84 (t, J= 5.6 Hz, 2H), 3.71 (s, 6H), 3.65-3.67 (m, 21H), 3.54-3.42 (m, 4H), 3.42- 3.41 (m, 2H), 3.31-3.28 (m, 2H), 2.69 (t, J= 6 Hz, 2H), 2.48 (t, J= 6 Hz, 2H), 1.43 (s, 9H).
di-tert-butyl 2,2'-((((5-(2,2-dimethyl-4,20-dioxo-3,8,11,14,17,24,27,30,33-nonaoxa-5,21-diazahexatriacontan-36-amido)isophthaloyl)bis(azanediyl))bis(ethane-2,1-diyl))bis(([1,1'-biphenyl]-4-ylsulfonyl)azanediyl))(2S,2'S)-bis(3-methylbutanoate)(15). Amide 15 was synthesized from amine 4 (92 mg, 0.1680 mmol, 2 eq) and dicarboxylic acid 14 (64 mg, 0.084 mmol, 1 eq) in dry DMF (2 mL) following the general procedure. The crude was purified by flash chromatography using an Isolute cartridge (SI 5g) (in gradient from 100% CHCl3 to CHCl3 :MeOH 50:1) affording the pure compound as a colorless oil (66 mg, 46% yield).1H NMR (400 MHz, CDCl3) δ: 9.08 (s, 1H), 8.22 (s, 2H), 8.02 (bs, 1H), 7.93-7.91 (m, 4H), 7.55-7.53 (m, 4H), 7.47-7.42 (m, 8H), 6.99 (bs, 1H), 3.93-3.92 (m, 4H), 3.84 (t, J= 5.6 Hz, 2H), 3.77-3.61 (m, 40H), 3.33-3.27 (m, 4H), 2.67 (t, J= 5.2 Hz, 2H), 2.49 (t, J= 6 Hz, 2H), 1.42 (s, 9H), 1.24 (s, 18H), 1.05, 0.89 (2xd, J= 6.4 Hz, each 6H).
General procedure for the TFA-mediated acid hydrolysis for the synthesis of compounds 7, 2, 12, 3
To a solution of the appropriate NH-Boc or tBu protected derivative (1 eq) in DCM (20 mL/mmol), TFA (30-342 eq) was added dropwise at 0°C. After completion of the reaction monitored by TLC, the solvent was co-evaporated with toluene (3x) and Et2O (3x).
(S)-19-([1,1'-biphenyl]-4-ylsulfonyl)-20-carboxy-21-methyl-15-oxo-3,6,9,12-tetraoxa-16,19-diazadocosan-1-amine trifluoroacetate salt (7). The title compound was prepared from NH-Boc-protected derivative 6 (180 mg, 0.235 mmol) treated dropwise with TFA (0.5 mL, 30 eq). The reaction was stirred for 1h at 0°C and then treated following the general procedure. The crude residue was purified by flash chromatography using an Isolute cartridge (SI 10g) (CHCl3:MeOH 50:1) to give compound 7 as a colorless oil (130 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ: 7.94-7.90 (m, 2H), 7.89-7.87 (m, 2H), 7.77 (bs, 1H), 7.71-7.68 (m, 2H), 7.58-7.56 (m, 2H), 7.52-7.46 (m, 3H), 7.43-7.39 (m, 1H), 3.77 (d, J= 6 Hz, 1H), 3.77-3.75 (m, 2H), 3.73-3.45 (m, 18H), 3.22 (bs, 2H), 2.65-2.64 (m, 2H), 2.13-2.07 (m, 1H), 1.23 (s, 9H), 1.02, 0.95 (2xd, J= 6.8 Hz, each 3H). 19F NMR (376 MHz, CDCl3) δ: -75.9.
Trimeric derivative 2. The title compound was prepared from tert-butyl ester 9 (47 mg, 0.021 mmol) treated dropwise with TFA (0.56 mL, 342 eq). The reaction was stirred for 15h at rt and then treated according to the general procedure to give final compound 2 as a white solid (45 mg, quantitative yield) after trituration with n-hexane. Mp: 70-73°C; 1H NMR (400 MHz, acetone-d6) δ: 8.49 (s, 3H), 7.95-7.93 (m, 6H), 7.84-7.82 (m, 6H), 7.72-7.70 (m, 6H), 7.52-7.48 (m, 6H), 7.44-7.41 (m, 3H), 4.07 (d, J= 6 Hz, 3H), 3.66-3.51 (m, 64H), 3.31-3.23 (m, 4H), 2.37 (t, J= 6 Hz, 6H), 2.23-2.12 (m, 3H), 1.02, 0.97 (2xd, J= 6.8 Hz, each 9H). 13C NMR (100 MHz, acetone-d6) δ: 172.0, 166.6, 145.8, 139.7, 138.9, 135.9, 129.8, 129.4, 129.2, 128.9, 128.0, 127.9, 71.0, 70.9, 70.8, 70.6, 70.2, 67.6, 66.6, 44.8, 40.5, 40.4, 37.1, 20.3, 19.6. HRMS (ESI, m/z) calculated for C99H135N9O30S3 [M−H]−: 2024.84042; found: 2024.83972. Elemental analysis calcd (%) for C99H135N9O30S3 : C 58.65, H 6.71, N 6.22; found: C 58.71, H 6.75, N 6.25.
15-((3,5-bis(methoxycarbonyl)phenyl)amino)-15-oxo-3,6,9,12-tetraoxapentadecan-1-amine trifluoroacetate salt (12): The title compound was prepared from NH-Boc-protected compound 11 (320 mg, 0.5608 mmol) treated dropwise with TFA (1.3 mL, 30 eq). The reaction mixture was stirred for 3h at 0°C and then treated following the general procedure to give the trifluoroacetate salt amine 12 as a white solid. 1H NMR (400 MHz, CDCl3) δ: 9.38 (bs, 1H), 8.44-8.43 (m, 2H, Ar), 8.41-8.40 (m, 1H, Ar), 7.53 (bs, 3H), 3.93 (s, 6H), 3.88 (t, J= 5.6 Hz, 2H), 3.78-3.76 (m, 2H) 3.69-3.59 (m, 12H), 3.17-3.13 (m, 2H) 2.80 (t, J= 5.6 Hz, 2H). 19F NMR (376 MHz, CDCl3) δ: -75.9.
(2S,2'S)-2,2'-((((5-(2,2-dimethyl-4,20-dioxo-3,8,11,14,17,24,27,30,33-nonaoxa-5,21-diazahexatriacontan-36-amido)isophthaloyl)bis(azanediyl))bis(ethane-2,1-diyl))bis(([1,1'-biphenyl]-4-ylsulfonyl)azanediyl))bis(3-methylbutanoic acid)(3): The title compound was prepared from compound 15 (75 mg, 0.047 mmol) treated dropwise with TFA (0.52 mL, 144 eq). The reaction was stirred for 5h at rt and then treated following the general procedure to give the trifluoroacetate salt amine 3 (60 mg, 83%yield) as a white semisolid after trituration with Et2O. 1H NMR (400 MHz, CD3OD) δ: 8.17 (d, J= 1.2 Hz, 1H), 7.98-7.94 (m, 4H, Ar), 7.96-7.94 (m, 4H, Ar), 7.64-7.62 (m, 4H), 7.47-7.38 (m, 7H), 4.09 (d, J= 10.4 Hz, 2H), 3.93-3.46 (m, 48H), 3.11 (t, J= 5.6 Hz, 2H), 2.66 (t, J= 5.6 Hz, 2H), 2.45-2.42 (m, 2H), 1.06, 1.02(2xd, J= 6.4 Hz, each 6H); 13C NMR (100 MHz, CD3OD) δ:172.8, 172.7, 171.2, 167.5, 145.5, 139.1, 137.9, 135.1, 128.7, 127.9, 127.1, 126.8, 121.5, 120.9, 70.1, 70.0, 69.9, 69.8, 69.7, 69.4, 66.8, 66.4, 43.5, 40.2, 39.1, 38.9, 37.2, 35.9, 28.5, 19.3, 18.5. 19F NMR (376 MHz, CDCl3) δ: -76.9. HRMS (ESI, m/z) calculated for C68H93N7O20S2 free base [M−H]−: 1390.58440; found: 1390.58447. Elemental analysis calcd (%) for C70H94F3N7O22S2 : C 55.80, H 6.29, N 6.51; found: C 55.83, H 6.31, N 6.56.
Synthesis of 5-(2,2-dimethyl-4,20-dioxo-3,8,11,14,17,24,27,30,33-nonaoxa-5,21-diazahexatriacontan-36-amido)isophthalic acid (14)
To a solution of the dimethyl ester 13 (213 mg, 0.27 mmol) in 6 mL of MeOH, a solution of LiOH 1N (2.6 mL) was added. The reaction was stirred for 3 h and then evaporated under reduced pressure. The crude was dissolved in water and the solution was acidified up to pH<2. The acid solution was extracted with EtOAc (3x) and the combined organic extracts were dried over Na2SO4, filtered and evaporated to give compound 14 as a white semisolid (165 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ: 13.15 (bs, 2H), 10.30 (s, 1H), 8.44-8.43 (m, 2H, Ar), 8.14-8.13 (m, 1H), 7.87 (m, 1H), 6.71 (bs, 1H), 3.71 (t, J= 5.6 Hz, 2H), 3.57-3.36 (m, 30H), 3.17-3.15 (m, 2H), 2.30 (m, t J= 5.6 Hz, 2H), 1.36 (s, 9H)
Biological assays
Enzyme inhibition assays.
Human proMMP-9 was produced by recombinant expression in Sf9 insect cells and purified by gelatin-Sepharose chromatography. Next, proMMP-9 was activated by incubation with the catalytic domain of MMP-3 (cat. No. 444217, Merck Millipore, Darmstadt, Germany) as previously described [43]. Recombinant human proMMP-1 and proMMP-2 were purchased from R & D systems (Minneapolis, MN, USA), dissolved in assay buffer (150 mM NaCl, 5 mM CaCl2, 0.01% Tween-20, 50 mM Tris, pH 7.4) to a concentration of 100 µg/mL and activated by incubation with 1 mM p-aminophenylmercuric acetate for, respectively, 4h and 1h. To measure MMP-1, MMP-2 and MMP-9 inhibition, the activated MMPs were incubated with different concentrations of inhibitors and incubated for 30 min at 37 °C. Next, the OmniMMP substrate peptide (Mca-PLGL-Dpa-AR-NH2, cat. no. BML-P126-0001, Enzo Life Sciences, Farmingdale, NY, USA) at a final concentration of 2.5 µg/mL was added and the increase in fluorescence over time was measured with the CLARIOstar microplate reader (BMG Labtech, Ortenberg, Germany). Proteolytic activity was determined by linear regression of the fluorescence curve and the percentage inhibition was calculated through comparison with the positive control (no inhibitors).
Recombinant human proMMP-12 was purchased from R&D Systems (Minneapolis, MN, USA). Pro-MMP-12 was auto activated by incubating in fluorometric assay buffer (FAB: Tris 50 mM, pH 7.5, NaCl 150 mM, CaCl2 10 mM, Brij-35 0.05%, and DMSO 1%) for 30 h at 37 °C. For assay measurements, the inhibitor stock solutions (DMSO, 10 mM) were further diluted in FAB. Activated enzyme (final concentration of 2.3 nM) and inhibitor solutions were incubated in the assay buffer for 3 h at 25 °C. After the addition of 200 μM solution of the fluorogenic substrate Mca-Lys-Pro-Leu-Gly-Leu-Dap(Dnp)-Ala-Arg-NH2 (Bachem) in DMSO (final concentration of 2 μM), the hydrolysis was monitored every 10 s for 15 min, recording the increase in fluorescence (λex = 325 nm, λem = 400 nm) with a Molecular Devices SpectraMax Gemini XPS plate reader (Molecular Devices, Sunnyvale, CA, USA). The assays were performed in triplicate in a total volume of 200 μL per well in 96-well microtiter plates (Corning black, NBS). Control wells lack inhibitor. The MMP inhibition activity was expressed in relative fluorescent units (RFU). Percent of inhibition was calculated from control reactions without the inhibitor. IC50 was determined using the formula vi/v0 = 1/(1 + [I]/IC50), where vi is the initial velocity of substrate cleavage in the presence of the inhibitor at concentration [I] and vo is the initial velocity in the absence of the inhibitor. Results were analyzed using SoftMax Pro software version 5.4.3 (Molecular Devices, Sunnyvale, CA, USA) and Prism Software version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Cell-based assays.
Cell culture
Human intestinal epithelial Caco-2 cell line were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 20% fetal bovine serum (FBS), 2 mM glutamine and 100 unit/ml penicillin-streptomycin at 37°C in a humidified atmosphere of 5 % CO2. Caco-2 cells were kindly donated by Prof Guido Bocci (Department of clinical and Experimental Medicine, University of Pisa).
Cell treatments
Caco-2 cells were seeded at density of 5x105 cells in 6-well plate and treated for 48h with lipopolysaccharide (LPS, 10 μg/mL, Sigma-Aldrich) and palmitate (PA, 400 µM, Sigma-Aldrich) to mimic the in vivo features of HFD exposure, in the absence or in the presence of MMP-12 inhibitors 1-3 (50 nM, 100 nM). Controls were run in parallel. Concentrations of PA and LPS were selected in accordance with previous reports [44–48]. Concentrations of compounds 1, 2 and 3 were selected based on IC50 and dose-finding experiments were performed to select the two doses more effective in counteracting the reduction of TJ proteins and IL-1β levels. After 72h, Caco-2 cells were lysed and culture media were collected.
Western blot assays
Cells were lysed as previously described [49,50] and proteins were quantified using Bradford assay. Proteins were separated on a pre-cast 4–20% polyacrylamide gel (Mini-PROTEAN® TGX gel, Biorad) and transferred to PVDF membranes (Trans-Blot® TurboTM, PVDF Transfer packs, Biorad). Then membranes were blocked with 3% BSA diluted in TBS (20 mM Tris-HCl, pH 7.5, 150 mM NaCl) with 0.1% Tween 20. Primary antibodies against GAPDH (#5174s, Cell Signaling), ZO-1 (Ab96587, Abcam) and claudin-1 (ab15098, Abcam) were used. Secondary antibodies were obtained from Abcam (anti-mouse ab97040 and anti-rabbit ab6721). Protein bands were detected with chemiluminescent ECL reagents (Clarityᵀᴹ Western ECL Blotting Substrate, Biorad). For the densitometry analysis, iBright Analysis software was used.
Assessment of interleukin (IL)-1β
The release of IL-1β from Caco2 cells was measured by ELISA kits (Abcam). Culture media were centrifuged for 5 min at 800 rpm to obtain cell-free supernatants. Supernatants (150 mL) were then used. Absorbance was expressed in percentage versus respective control.
Statistical analysis
The results are presented as mean ± S.E.M. of at least 3 independent experiments. The significance of differences was evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. P values <0.05 were considered significantly different. All statistical procedures were performed by commercial software (GraphPad Prism, version 7.0 from GraphPad Software Inc., San Diego, CA, USA).
{kind=link}
{kind=link}