Ethics statement
This study was performed in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Texas Biomedical Research Institute (permit number: 1419-MA).
Parasite lifecycle maintenance and recovery of Schistosoma mansoni worms
The S. mansoni lifecycle spans approximately 75 days (30 days development within snails and 45 days in hamsters). To safeguard against the loss of parasite populations, we establish duplicate cohorts of hamster infections ~ 3–4 weeks apart. Many of the same shedding snails are used to infect the two cohorts of hamsters. Hence, the parallel populations of each line form a single population, and some of our adult worm pools are collected one month apart.
To recover adult worms, we perfused infected Golden Syrian hamsters used for schistosome life cycle maintenance as previously described [19]. Briefly, we euthanized each hamster with a solution of 1 ml of phenobarbital (Fatal Plus) + 10% heparin and dissected the animal to expose the liver. We disrupted the hepatic portal vein using a needle and perfused the heart and the liver for around 1 minute each with a perfusion solution (193 nM of NaCl / 1mM EDTA) at a flow rate of 40 ml/minute using a peristaltic pump. After perfusion, we rinsed the intestine with normal saline and collected worms trapped in the intestine. All expelled worms were collected in a fine mesh sieve and rinsed with normal saline solution (154 nM of NaCl, pH 7.5). We then transferred the collected S. mansoni worms to a petri dish for counting and separation by sex. The worms were stored in 1.5 ml microcentrifuge tubes, flash-frozen in liquid nitrogen, and preserved at -80°C until gDNA extraction.
Cercarial shedding
We used datasets from Le Clec’h et al. [17] from 2015 and performed a similar infection experiment to measure cercarial production of SmBRE in 2023. Briefly, we exposed 240 BgBRE snails to a single SmBRE miracidium in 24-well plates overnight. We then transferred the exposed snails to trays for 4 weeks. At four weeks post-exposure, each snail was individually placed in a well of a 24 well-plate in 1 mL freshwater and kept under artificial light for 2 h to induce cercarial shedding. For each well with cercariae, we sampled three 10 µL (for the high shedder parasites) or 100 µL (for the low shedder parasites) aliquots and added 20 µl of 20× normal saline. We then counted the immobilized cercariae in triplicate under a microscope. We multiplied the mean of the triplicated measurement by the dilution factor to determine the number of cercariae produced by each infected snail. We monitored cercarial production weekly from week 4 to 7 post-exposure in SmBRE-infected snails. To track cercarial production of individual snails throughout the 4-week patent period, we isolated each infected snail in a uniquely labeled 100 mL glass beaker filled with ~ 50 mL freshwater at the first shedding. All snails were fed ad libitum with fresh lettuce and kept in the dark in the 26–28°C temperature-controlled room.
gDNA extraction, gDNA Library preparation and sequencing
We extracted gDNA from 27 to 100 single-sex worms per pool (Table 1) with the DNeasy Blood & Tissue Kit (Qiagen, Germantown, MD, USA), following the manufacturer protocol. We ground the worms in 180 µl of ATL buffer using a sterile micro pestle and added 20 µl of proteinase K before incubation at 56°C for 2h. gDNA was eluted in 75 µL of elution buffer. We quantified extracted gDNA using Qubit dsDNA BR Assay Kit (Invitrogen, Carlsbad, CA, USA) and performed library preparation using the KAPA Hyperplus Kit (Roche, Indianapolis, IN, USA) with 400 ng of input material. We used the manufacturer’s instructions with the following modifications for our library construction: enzymatic fragmentation time: 20 minutes, library amplification: six PCR cycles, library size selection: a first size cut at 0.6X (30 µl beads), and a second size cut at 0.8X (10 µl beads). The library sizes were assessed using TapeStation 4200 D1000 ScreenTape (Agilent, Santa Clara, CA, USA), and all libraries were quantified using the KAPA Library Quantification Kit (Roche, Indianapolis, IN, USA). Pooled libraries were submitted to Admera Health and sequenced to high read depth on a NovaSeq X Plus platform (Illumina) with 150 bp paired-end reads.
Table 1
| SampleID | Population | Collection Date | Pool size | Sex | Mean coverage | Coverage > 10X | Coverage < 1X | Accession1 |
| BRE110916_m | SmBRE | 11/09/16 | 64 | males | 49.5 | 96.3% | 1.9% | SAMN40565564 |
| BRE110916_f | SmBRE | 11/09/16 | 71 | females | 52.3 | 96.7% | 2.3% | SAMN40565565 |
| LE110216_m | SmLE | 11/02/16 | 77 | males | 48.8 | 96.4% | 1.9% | SAMN40565566 |
| LE110216_f | SmLE | 11/02/16 | 67 | females | 55.4 | 97.8% | 1.4% | SAMN40565567 |
| BRE062718_m | SmBRE | 06/27/18 | 98 | males | 69.4 | 96.8% | 2.0% | SAMN40565568 |
| BRE062718_f | SmBRE | 06/27/18 | 78 | females | 57.3 | 97.6% | 1.5% | SAMN40565569 |
| LE062118_m | SmLE | 06/21/18 | 86 | males | 61.5 | 96.4% | 2.2% | SAMN40565570 |
| LE062118_f | SmLE | 06/21/18 | 87 | females | 50.0 | 96.6% | 2.7% | SAMN40565571 |
| BRE051420_m | SmBRE | 5/14/2020 | 95 | males | 62.8 | 96.2% | 1.7% | SAMN40565572 |
| BRE051420_f | SmBRE | 5/14/2020 | 76 | females | 88.0 | 97.8% | 1.2% | SAMN40565573 |
| LE051420_m | SmLE | 5/14/2020 | 32 | males | 61.6 | 96.6% | 1.3% | SAMN40565574 |
| LE051420_f | SmLE | 5/14/2020 | 30 | females | 51.6 | 97.5% | 1.6% | SAMN40565575 |
| BRE112320_m | SmBRE | 11/23/2020 | 103 | males | 60.0 | 96.0% | 2.1% | SAMN40565576 |
| BRE112320_f | SmBRE | 11/23/2020 | 90 | females | 56.7 | 95.7% | 3.3% | SAMN40565577 |
| LE112320_m | SmLE | 11/23/2020 | 68 | males | 187.7 | 97.3% | 1.2% | SAMN40565578 |
| LE112320_f | SmLE | 11/23/2020 | 106 | females | 56.3 | 97.4% | 1.8% | SAMN40565579 |
| BRE070521_m | SmBRE | 7/5/2021 | 100 | males | 65.7 | 96.4% | 2.2% | SAMN40565580 |
| BRE070521_f | SmBRE | 7/5/2021 | 57 | females | 62.9 | 97.6% | 1.3% | SAMN40565581 |
| LE070521_m | SmLE | 7/5/2021 | 75 | males | 53.7 | 95.2% | 3.1% | SAMN40565582 |
| LE070521_f | SmLE | 7/5/2021 | 73 | females | 54.1 | 96.7% | 2.5% | SAMN40565583 |
| BRE122121_m | SmBRE | 12/21/2021 | 101 | males | 66.9 | 97.7% | 1.5% | SAMN40565584 |
| BRE122121_f | SmBRE | 12/21/2021 | 83 | females | 53.3 | 98.3% | 1.1% | SAMN40565585 |
| LE122121_m | SmLE | 12/21/2021 | 101 | males | 83.9 | 97.5% | 1.8% | SAMN40565586 |
| LE122121_f | SmLE | 12/21/2021 | 104 | females | 68.1 | 97.9% | 1.3% | SAMN40565587 |
| BRE070522_m | SmBRE | 7/5/2022 | 93 | males | 76.2 | 97.6% | 1.8% | SAMN40565588 |
| LE070522_m | SmLE | 7/5/2022 | 64 | males | 69.4 | 96.6% | 1.9% | SAMN40565589 |
| LE070522_f | SmLE | 7/5/2022 | 51 | females | 61.8 | 97.0% | 2.2% | SAMN40565590 |
| BRE021523_m | SmBRE | 2/15/2023 | 96 | males | 93.3 | 97.5% | 1.7% | SAMN40565591 |
| BRE021523_f | SmBRE | 2/15/2023 | 96 | females | 54.6 | 97.6% | 1.9% | SAMN40565592 |
| LE021523_m | SmLE | 2/15/2023 | 106 | males | 66.8 | 97.7% | 1.7% | SAMN40565593 |
| LE021523_f | SmLE | 2/15/2023 | 33 | females | 55.9 | 94.5% | 4.8% | SAMN40565594 |
| BRE092921_m | SmBRE | 9/29/2021 | 100 | males | 90.8 | 96.8% | 2.1% | SAMN40565595 |
| BRE092921_f | SmBRE | 9/29/2021 | 94 | females | 66.1 | 96.8% | 2.1% | SAMN40565596 |
| LE092921_m | SmLE | 9/29/2021 | 27 | males | 71.1 | 96.3% | 1.7% | SAMN40565597 |
| LE092921_f | SmLE | 9/29/2021 | 100 | females | 66.5 | 97.6% | 1.6% | SAMN40565598 |
| BRE102621_m | SmBRE | 10/26/2021 | 100 | males | 79.5 | 97.2% | 1.0% | SAMN40565599 |
| BRE102621_f | SmBRE | 10/26/2021 | 54 | females | 98.0 | 98.5% | 1.0% | SAMN40565600 |
| LE102621_m | SmLE | 10/26/2021 | 60 | males | 93.7 | 97.0% | 1.9% | SAMN40565601 |
| 1All accession numbers are in bioproject PRJNA1090435 | | | | |
Computational environment
We used conda v23.1.0 to manage environments and download packages required for the analysis. Data processing was performed in R 4.2.0 using tidyverse v1.3.2, and figures were generated with ggplot v3.4.2.
Genotyping
We used trim_galore v0.6.7 [20] (-q 28 --illumina --max_n 1 --clip_R1 7 --clip_R2 7) for adapter and quality trimming before mapping the sequences to version 10 of the S. mansoni reference genome
(Wellcome Sanger Institute, BioProject PRJEA36577) with BWA v0.7.17-r118 [21] and the default parameters. We used GATK v4.3.0.0 [22] for further processing of the sequences. First, we removed all optical/PCR duplicates with MarkDuplicates. Next, we used HaplotypeCaller and GenotypeGVCFs to call single nucleotide variants (SNV) on a contig-by-contig basis. These were aggregated for each pooled sample and further consolidated into a comprehensive VCF file encompassing all sequences. Quality filtering was performed using VariantFiltration with recommended parameters (FS > 60.0, SOR > 3.0, MQ < 40.0, MQRankSum < -12.5, ReadPosRankSum < -8.0, QD < 2.0). Additionally, we used VCFtools v0.1.16 [23] for refining, specifically excluding non-biallelic sites with quality < 15 and read depth < 10, along with sites and individuals with a genotyping rate < 50%.
We measured selection coefficient (s) at each SNP locus by fitting a linear model between the natural log of the allele ratio (freq[allele1]/freq[allele2]) against generation time (measured as the number of 75-day parasite life cycles). The raw s values were smoothed by computing the running medians to remove noise.
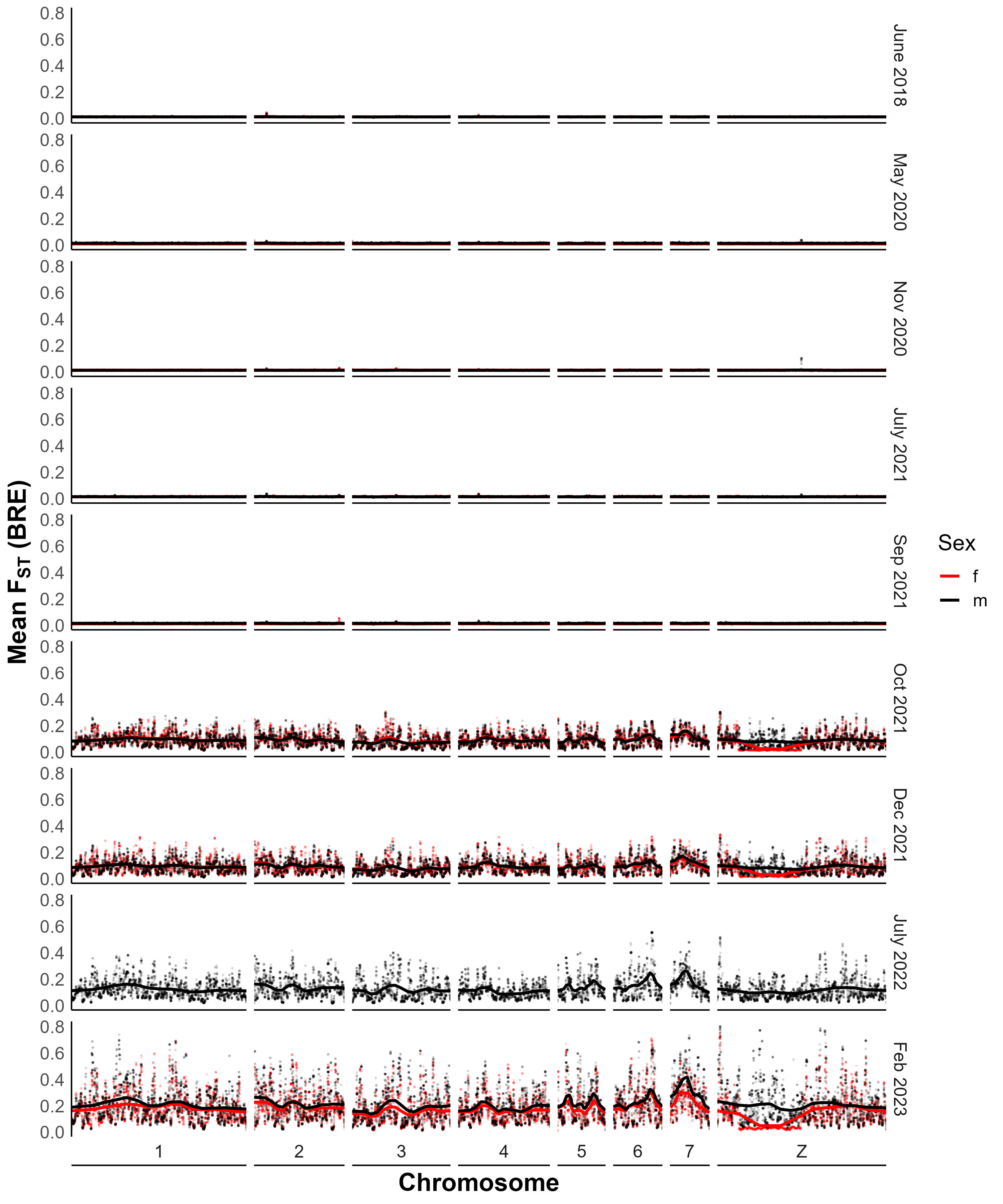
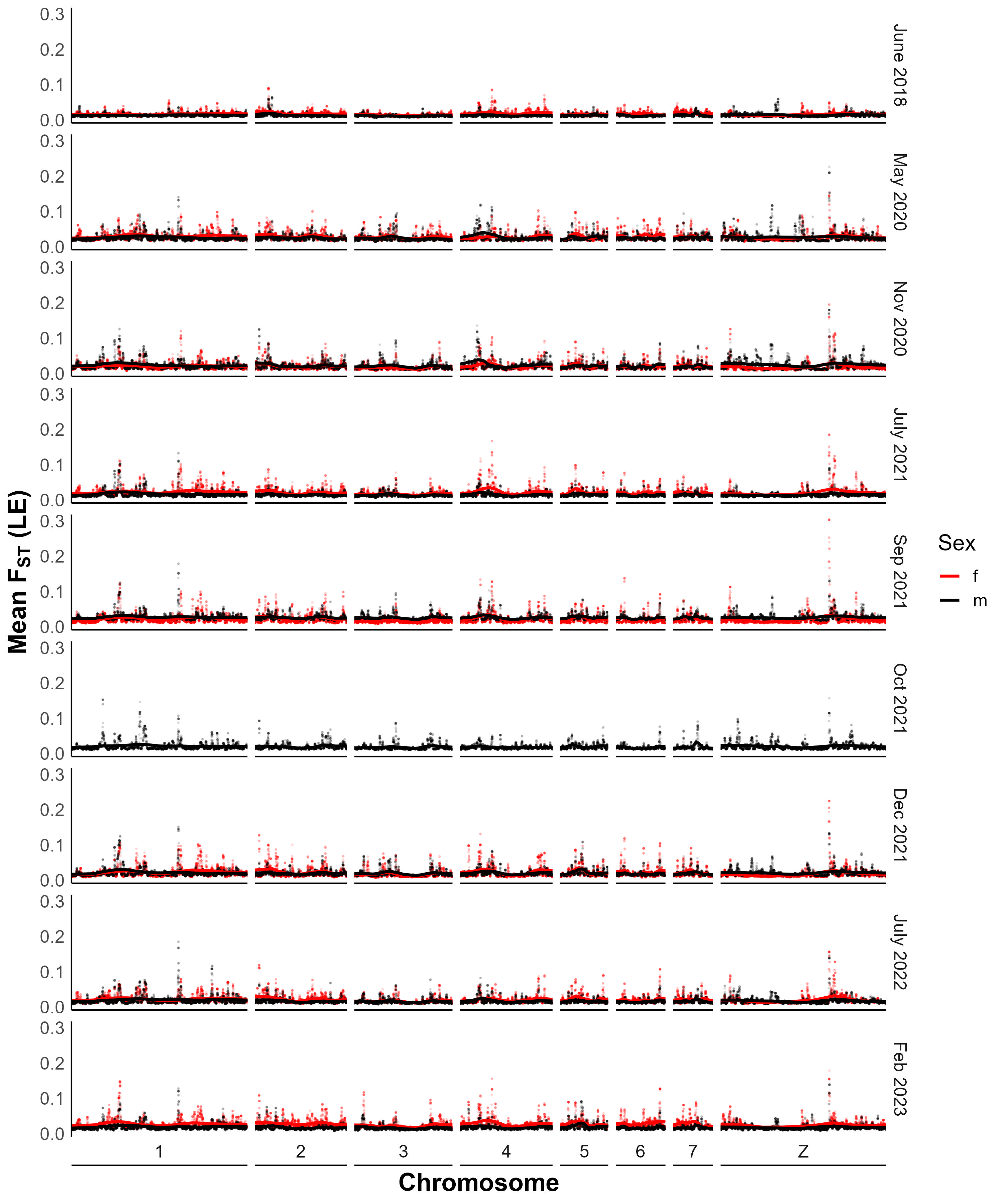
FST statistics
We calculated FST with popoolation2 [24], a pipeline designed for analysis of pooled samples. Briefly, we used samtools v1.9 [25] mpileup to generate a joint bam file containing sequences from two different samples to make comparisons across time or between populations. Next, we converted the file to a suitable input file for popoolation2 with mpileup2sync.jar, keeping only bases with a minimum quality of 20. Finally, we calculated FST with fst-sliding.pl and the following parameters: “--suppress-noninformative”, “--min-count 6”, “--min-coverage 50”, “--max-coverage 200”, “--min-covered-fraction 1”, “--window-size 1”, “--step-size 1”, and the relevant pool sizes with “--pool-size.” We then calculated mean FST in 20 kb windows using a custom function in R and added the smoothing line using the locfit method from the locfit v1.5-9.8 package.
To calculate FST for SmLE specific variants, we modified the parameters above to “--min-coverage 10” and “--max-coverage 6000” and overlapped the resulting files with known variant loci.
Statistical analysis
We performed all statistical analyses with the rstatix v0.7.2 package [26]. For normally distributed data (Shapiro test, p > 0.05), we performed parametric Student's t-test to compare time points. Otherwise, we used non-parametric Wilcoxon rank-sum tests. We adjusted p-values for multiple comparisons using the Benjamini–Hochberg method when needed and considered these significant when p < 0.05 [27].
{kind=link}
{kind=link}