The central importance of Triatominae in the transmission of Chagas disease has driven the need for phylogenetic studies to better characterize and incriminate Triatominae species, and the establishment of laboratory colonies to undertake in vivo experiments addressing questions on vector biology, vector-parasite interactions, insecticide susceptibility, among others. Despite the generation of considerable molecular resources for Triatominae, such as genome and genome expression data, the virome infecting Triatominae remains poorly known. The first virus discovered in Triatominae was the TrV, isolated from wild and laboratory populations of T. infestans from Argentina in 1987 [7, 8] but it was not until 2000 that the first TrV genome (GenBank: NC_003783) became publicly available [32]. Only recently, a further seven viruses belonging to the families Iflaviridae, Permutotetraviridae and Solemoviridae (RpV1–7; GenBank: MZ328304–310) have been detected in laboratory colonies of R. prolixus from Brazil [16]. One other virus—MlV1, belonging to the family Iflaviridae—has been identified in Meccus longipennis from Mexico (GenBank: OQ384707). Crucially, only a single reference genome exists for each virus species so far detected in Triatominae. In this study, we sought to assess the utility of publicly available transcriptome data available through the SRA and ENA repositories for better characterizing Triatominae viromes, assembling additional and novel virus genomes and establishing new virus records for a variety of species and genera in Triatominae.
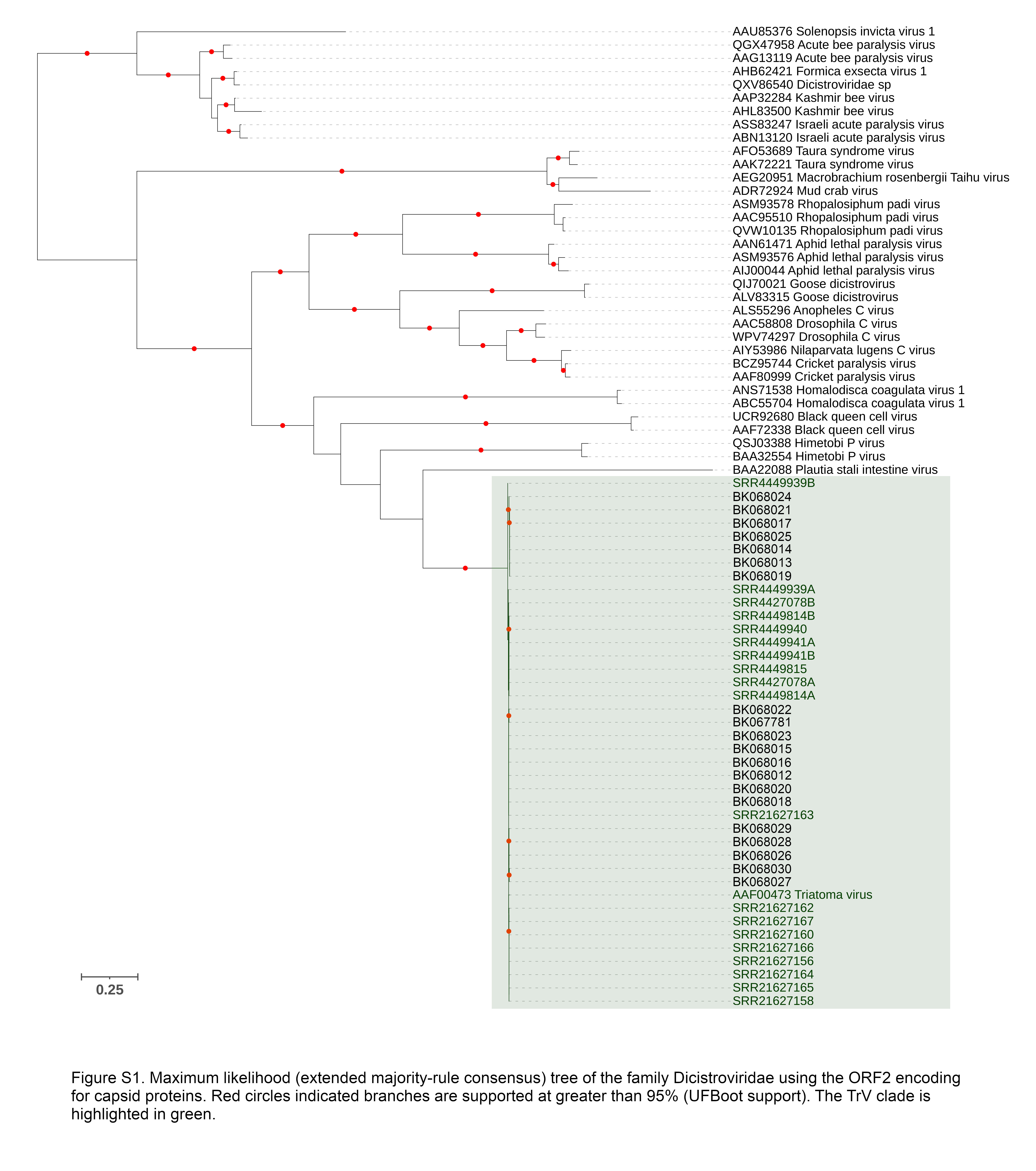
The TrV was the most prevalent virus detected among samples, species and genera. Previously, this virus was detected in a variety of Triatoma species as well as P. coreodes and R. prolixus. To our knowledge, this study reports the first records of TrV in T. brasiliensis and in the genus Mepraia (M. gajardoi, M. spinolai, M. parapatrica). We have expanded the publicly available genomic resources of TrV, adding 39 genomes to the single genome currently available on GenBank. No clear geographic partitioning of genetic variation between laboratory and wild host populations was apparent. The TrV has previously been detected in several Triatoma species in Argentina, with positivity rates of approximately 10% [11, 13, 14], whereas positivity rates among laboratory Triatominae have been reported to be considerably higher, in some cases up to 100% [14]. This has been attributed to high host densities and the increased likelihood of cannibalism, coprophagy, kleptohaemophagy, and fecal-oral transmission [14, 33]. However, in the current study, we assembled partial and whole TrV genomes from almost 50% of Mepraia individuals (8/18) collected from Chile as part phylogenomic study of the genus [34], and from 100% of seven T. infestans individuals collected from Bolivia as part of a study to establish expression profiles for the species [35]. Far fewer TrV positives (1 of 29) were detected among individuals taken from a wild population of T. brasiliensis from the city of Caicó, Brazil as part of an effort to establish a reference transcriptome and expression profiles for this species [36, 37]. While TrV seropositivity has been found among human populations in Argentina, Bolivia, and Mexico [38], to date the virus has only detected among wild Triatominae populations in Argentina. To our knowledge, our study reports the first cases of TrV detected among wild populations in Bolivia, Brazil, and Chile. Taken as a whole, the TrV appears to have a continental distribution and considerable differences in prevalence among wild Triatominae, with some populations reaching the levels of prevalence found in laboratory populations. Due to the virus’ horizontal mode of transmission and high pathogenicity, it has been considered a potential agent for the control of Triatominae vectors [14]. The considerable variation in prevalence observed in wild populations may influence host exposure, susceptibility, and immune response, which have important implications for the efficacy of vector control measures using this agent. The impact of the TrV genomes with ORF1 stop codons, and whether they can be rescued with functional virus proteins in the infected cells, needs further elucidation. The genomic variation extant in endemic and introduced TrV may also have an important influence the efficacy of such control interventions. Our contributions to expanding the public genomic resources of this virus serves as an important baseline to further characterize genomic and geographic variation in TrV.
The next most prevalent virus among Triatominae hosts was MlV1. This virus is very poorly known and first characterized in 2023 from wild caught individuals of M. longipennis from the state of Chihuahua, Mexico. The MlV1 genomes assembled in the current study came from wild and laboratory populations, and the genetic variation and phylogenetic position is consistent with a single taxonomic clade. It is found in a sister relationship with a clade of other Hemiptera-specific viruses (Euscelidius variegatus virus 1, Psammotettix alienus iflavirus 1 and Graminella nigrifrons virus 1), which may represent host-based evolutionary relationships. Our findings present the first records of this virus both in laboratory populations, and from wild populations in South America.
The Drosophila melanogaster NV was detected in two individuals from a laboratory colony in Argentina. This virus was first characterized in a laboratory colony of Drosophila melanogaster in Sweden in 2006. Since then, the virus has been detected in species from a total of five insect Orders (Diptera, Coleoptera, Hymenoptera, Lepidoptera and Orthoptera) [39]. Our findings record Drosophila melanogaster NV in a species of Hemiptera (T. infestans) for the first time. Although the assemblies recovered in the current study display very low levels of sequence differences with the NV references available on GenBank, our assemblies are partial, representing less than 20% of the viral genome, and so we are unable to fully characterize the extent of differentiation between the virus found in the Argentinian colony from the Drosophila melanogaster NV references. However, our findings present a primer for further work on the detection and characterization of Drosophila melanogaster NV in Triatominae and its potential circulation among species across the Order.
The final group of viruses detected in this study were only recently characterized from a R. prolixus laboratory colony from Brazil [16]. Here we detected signatures for three of the seven originally characterized viruses in individuals of R. robustus and R. neglectus. These three viruses belong to the families Permutotetraviridae (RpV4) and Solemoviridae (RpV5, RpV6) and are the first reports of such detections outside of Brazil. We also report the first records of the Solemoviridae viruses detected in a wild Triatominae (R. robustus) population, which were collected in French Guiana as part of a study of chemosensory genes in Chagas disease vectors [40]. Whereas the study that originally characterized the R. prolixus viruses found that the persistence of these viruses in laboratory colonies was at least partly explained by the vertical modes of transmission [16], the modes of transmission in wild populations is unknown. Our findings indicate the circulation of R. prolixus viruses in wild populations of other Rhodnius species and the need for further exploration of these viruses, their transmission routes and pathogenic potential across the genus.
Understanding the relationship between insect-specific viruses and their Triatomine hosts may unlock novel population suppression mechanisms that can be leveraged in the fight against the silent expansion of the neglected, but deadly Chagas disease in the New World. All six of the viruses detected in the current study are now reported in well-established laboratory colonies of Triatominae (this study and Brito et al. [16]). The hidden presence of such insect viruses in vector colonies can have important impacts on the quality and reproducibility of downstream experimental studies of virology, immunology, and metabolism, where coinfections can inhibit the replication of a pathogen and alter vector competence, alter virus virulence and infection severity, and modify immune responses to primary and secondary infections [41–44]. Thorough screening of laboratory colonies using agnostic sequencing approaches enables detection and characterization of potentially hidden and novel virus infections and control for such variables that may impact experimental outcomes. Our study also underlines the importance of further virome studies in wild Triatominae populations. There is currently a dearth of publicly transcriptome data to enable mining of novel and diverse viruses from the wild. Despite minimal available data, our study has provided new virus records for each wild origin dataset analyzed, showing the values of reinterrogating publicly available transcriptome data for pathogen discovery. Consequently, we recognize continued agnostic sequencing approaches as highly valuable for establishing baselines for novel and emerging virus prevalence, diversity, and dynamics among Chagas disease vectors and essential for possible novel virus isolation and pathogenicity analysis.
{kind=link}