At present, the molecular imaging probes used for cardiac imaging indicate changes in myocardial perfusion25,26, ventricular structure and function27,28, and metabolism29–34. Changes in perfusion and myocardial function are associated with cardiac tissue remodeling and essentially indicate disease that is already evident by other imaging modalities35. By contrast, probes which report on changes in cardiac metabolism may be more suitable for detecting pre-symptomatic or sub-clinical disease as metabolic changes occur prior to the onset of cardiac injury5,36. Clinical cardiometabolic PET imaging is dominated by the use of [18F]FDG. [18F]FDG has the advantages of being readily available in most major hospitals and that its uptake in the heart correlates with some indices of cardiac dysfunction. Nevertheless, cardiac [18F]FDG PET imaging is subject to the potentially confounding effect of uptake of this probe by other metabolically active cells. For example, [18F]FDG, is avidly taken up by inflammatory cells and is not easily displaced from these cells. The result of this uptake pattern is decreased image sensitivity37. Moreover, the first step of glucose metabolism, which is shared by [18F]FDG, is its phosphorylation to glucose-6-phosphate. This molecule can undergo glycolysis to afford pyruvate or enter the pentose phosphate pathway. These divergent metabolic pathways may be active to different extents in the diseased heart and may explain the reports of spatial and temporal variability of [18F]FDG in hearts with cardiotoxicity38.
Agents which report on changes in fatty acid metabolism have the potential to directly assess the energetic state of the heart due to this tissue’s reliance on fatty acid oxidation for ATP. Some of the PET tracers developed for this application include [18F]fluoro-6-thia-heptadecanoic acid29,30, [11C]palmitate34, [11C]acetate31, and [11C]lactate32. Unfortunately, their imaging applications are limited either by poor retention in the heart or catabolism by other biochemical pathways which do not correlate with the incidence of cardiac disease29–34. Of the many alternative substrates used by the LCFA-deficient heart, SCFA are preferentially used as they exploit the existing mechanisms for lipid catabolism5,6. Therefore, we hypothesized that the SCFA analog, [18F]FPA, could be used as an indicator of cardiometabolic dysfunction caused by doxorubicin-induced cardiotoxicity.
We began our assessment of [18F]FPA as a cardiac imaging agent by analyzing its retention in fed and fasted states. Plasma concentrations of endogenous SCFAs may be influenced by fasting as they are predominantly sourced from the gut microbiome39. By contrast to [18F]FDG14,40, the tissue uptake of [18F]FPA is only marginally influenced by a 6 h fasting period. On this basis, we determined that fasting does not suppress cardiac uptake of [18F]FPA with the consequence that it is not required before image acquisition. These findings support the practicality of this imaging probe for assessing cardiac disease.
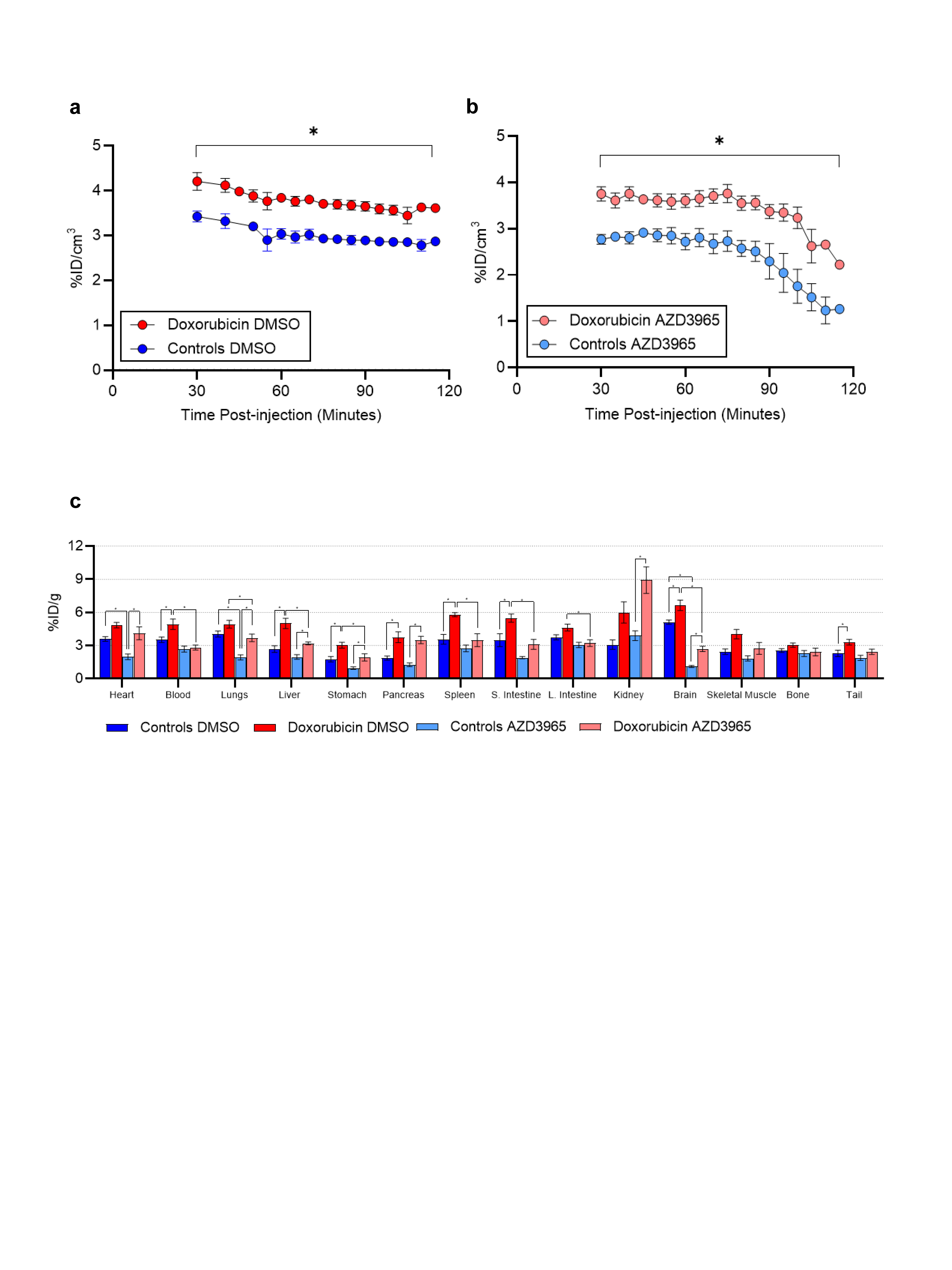
Our experiments in healthy mice indicate that [18F]FPA is taken up by nearly every tissue (Fig. 1), which we reasoned could diminish image contrast in the region of the heart and result in undesirable radiation doses to patients. Therefore, we enhanced the contrast to cardiac tissue by co-injecting [18F]FPA with AZD3965, an inhibitor of MCT141. We anticipated this combination would significantly reduce the uptake by non-cardiac tissues reliant on MCT1 for uptake of SCFA7,15,16. By contrast, SCFA freely enter cardiomyocytes without the need for these transporters7. The dose of AZD3965 (5 mg/kg) used to enhance cardiac image contrast may not directly translate to humans as it is several fold greater than the maximal recommended dose for patients (30 mg/70 kg)42. However, differences in the pharmacokinetics of this drug are apparent between humans and mice. For example, the therapeutic and subtoxic dose for mice is 100 mg/kg43,44. Since we suppressed background uptake of [18F]FPA at 20-fold lesser concentrations of AZD3965 in mice (Fig. 2A), it may be possible to titrate this drug for use in patients to doses substantially below 30 mg.
We tested our approach in a model of doxorubicin-induced cardiotoxicity because treatment of this condition would benefit from the identification of suitable imaging biomarkers for diagnosis and monitoring response to treatment. Cardiac dysfunction caused by doxorubicin is mechanistically and pathologically distinct from more common cardiac diseases which typically lead to cardiac hypertrophy and fibrosis35. Therefore, relying on standard physiological and anatomical markers for diagnosis may not be effective for identifying patients at risk of developing disease. [18F]FPA uptake was elevated in hearts exposed to doxorubicin and was correlated to increased activities of ACSS. This correlation suggests a mechanistic basis for the increased cardiac uptake of [18F]FPA due to doxorubicin-induced cardiac injury.
Doxorubicin induces several potentially pathological changes in mice and humans, including systemic inflammation, weight loss, and cardiotoxicity3,20. This complex mixture of changes is likely responsible for the increased uptake of [18F]FPA (Fig. 4) in blood and non-cardiac tissues. Encouragingly, co-administration of AZD3965 effectively decreased the signal due to [18F]FPA in all peripheral tissues of the doxorubicin-treated mice while maintaining cardiac uptake of [18F]FPA. Inflamed tissues are likely to use more SCFA as alternative substrates for metabolism or for promoting anti-inflammatory signaling cascades45. In this light, the ablation of doxorubicin-induced uptake of [18F]FPA in blood, bones, spleens, and tails (Supplementary Fig. 1C) may reflect inhibition of its uptake by inflammatory cells, which include myeloid populations in the bone, lymphocytes in the spleens, and a combination of both in the blood. In the tails, intravenous injections may be sufficient to stimulate activation of inflammatory cells in the blood which are already primed for activation by doxorubicin46. Doxorubicin exposure significantly increased brain uptake of [18F]FPA, but this was largely abolished by AZD3965 due to the requirement of MCT1 for permeability of SCFA through the blood brain barrier47,48. The term “chemobrain” is used to describe cognitive dysfunction that can arise during chemotherapy. Cognitive dysfunction is attributed to inflammatory and morphological changes which occur in the brain due to doxorubicin toxicity49. These events could be responsible for the increased [18F]FPA uptake in the brains of these animals and suggests a possibility for using this tracer to image brain health as it is exposed to doxorubicin. Further study is needed to determine the precise mechanisms which account for all the aforementioned changes in response to doxorubicin, but our observations suggest that AZD3965 renders [18F]FPA PET feasible even when systemic inflammation is present.
Interestingly, AZD3965 induces a progressive loss of cardiac signal beginning at 100 min p.i. which is not observed in the absence of the drug (Supplementary Fig. 1A). These decrements are proportionate in the hearts of both doxorubicin-treated and untreated controls (Supplementary Fig. 1B) and therefore do not invalidate our comparisons between these groups. The plasma half-life of AZD3965 is approximately 2.5 h44. One possible explanation for the decreasing signal is the accelerated clearance of [18F]FPA from blood by AZD3965, as decrements are also evident in other highly perfused tissues (Supplementary Fig. 1C). Nevertheless, the stable cardiac signal from 30–90 min p.i. affords a broad imaging window that can be readily implemented for clinical scans.
Although we employed a well-characterized model of doxorubicin-induced cardiotoxicity for the evaluation of cardiac [18F]FPA PET, we anticipate this imaging strategy may be useful in detecting other forms of cardiac injury. An early and pronounced shift in fatty acid metabolism is a common feature of all cardiac diseases7,50. Consequently, we plan to investigate [18F]FPA as a probe for detecting incipient cardiac failure across the spectrum of heart disease, not only the disease arising from cardiotoxicity. We anticipate that our approach will also be translatable to clinical populations. Prior human studies with [18F]FPA12 and AZD396542 in other indications confirm their safety.
One major limitation of our study is that we investigated [18F]FPA PET when cardiac dysfunction, as evidenced by decreased fractional shortening, was already evident. At this stage of disease, diagnostic imaging is possible using non-nuclear modalities and cardiac damage is irreversible. Therefore, our future work will investigate the utility of [18F]FPA PET as an early indicator of doxorubicin-induced cardiotoxicity. In addition, we found, as have others, that female C57BL/6 mice are not as susceptible to doxorubicin toxicity as males51. Although observations in clinical populations support the hypothesis that cardiometabolic changes occur similarly in men and women, we were unable to test this hypothesis in our model.
{kind=link}