Preparation of Cell culture
In the current study, we used murine macrophages (BMDMs and J774a.1 cells). Bone marrow derived macrophages (BMDMs) were obtained from femur and tibia of C57BL/6 mice as described previously [21]. In brief, cells were obtained from femur and tibia of 6 to 8 weeks old mice and cultured in RPMI1640 (Hyclone) medium supplemented with 10% FBS in the presence of 10 ng/ml M-CSF (Pepro Tech) and 1% streptomycin and penicillin (Gibco) in CO2 incubator at 37oC for 7 days. Fresh cell culture medium was added at day 4 of incubation and removed the dead or non-adherent cells. At day 8, the adherent BMDM cells were collected and transferred to 12-well cell culture plates for further experiments. J774a.1 cells were collected from cold storage -80oC (previously obtained from the Cell Culture Center, Xiehe Medical University (Beijing, China)) and cultured for 2 to 3 days in cell culture flasks in DMEM (Hyclone, Logan, UT, USA) medium in the presence of 10-15% FBS (Gibco, Grand Island, NY, USA), and 1% streptomycin and penicillin (Gibco) at 37oC in CO2 (5%) incubator [22]. After 2 to 3 days, J774a.1 cells were transferred into 12-well culture plates for further experiments.
Preparation of Bacterial culture
In our current experiment, we used virulent strain of M. bovis. M. bovis Beijing strain C68004 was obtained from China Institute of Veterinary Drug Control (CVCC, China). A stock culture of M. bovis was maintained in Middlebrook 7H9 medium (Difco) enriched with 10% albumin-dextrose-catalase (ADC), 2 mg/L sodium pyruvate and 0.05% Tween-80 at 37°C under biosafety conditions level 3 (BSL3). M. bovis was cultured for 2-3 weeks at 37oC to a concentration of 107-8/ml before used for cells or animal infection.
Mice model of M. bovis infection
C57BL/6 female mice (6-8 weeks of age) were obtained from Vital River Laboratories (Beijing, China). All mice were maintained in a strict biosafety measures in BSL-III laboratory of China Agricultural University, under the protocols of the Laboratory Animal Ethical Committee of China Agricultural University (Protocol 20110611-01). Mice were housed in groups as many as 5 mice per cage with free access to feed and water under sterilized condition. Mice were infected with M. bovis at 100 CFU (Colony-forming units) via intranasal (i.n) route. The suspension of M. bovis bacilli was prepared in sterilized PBS, the negative controlled animals were treated to PBS. Mice were properly anaesthetized by injecting PBS diluted Zoletil 50 (50mg/kg; Virbac, France) via intraperitoneal route [23] before infection with M. bovis. One day post infection (p.i), about five mice were selected randomly and sacrificed by cervical dislocation, under ethics requirements [24], as to calculate viable bacilli in the lung tissues. Next, 6 mice were randomly selected from each experimental group at 21 and 84 day post infection. Blood samples were collected from orbital route [25] of mice after proper anaesthetizing by intraperitoneal injection of PBS diluted Zoletil 50 (50mg/kg; Virbac, France). Serum samples were obtained from blood by low-speed centrifugation and tissues (lung, spleen) were collected aseptically from all experimental animals and stored at -80°C till further experiments.
In vivo IFNAR1 blocking antibody treatments
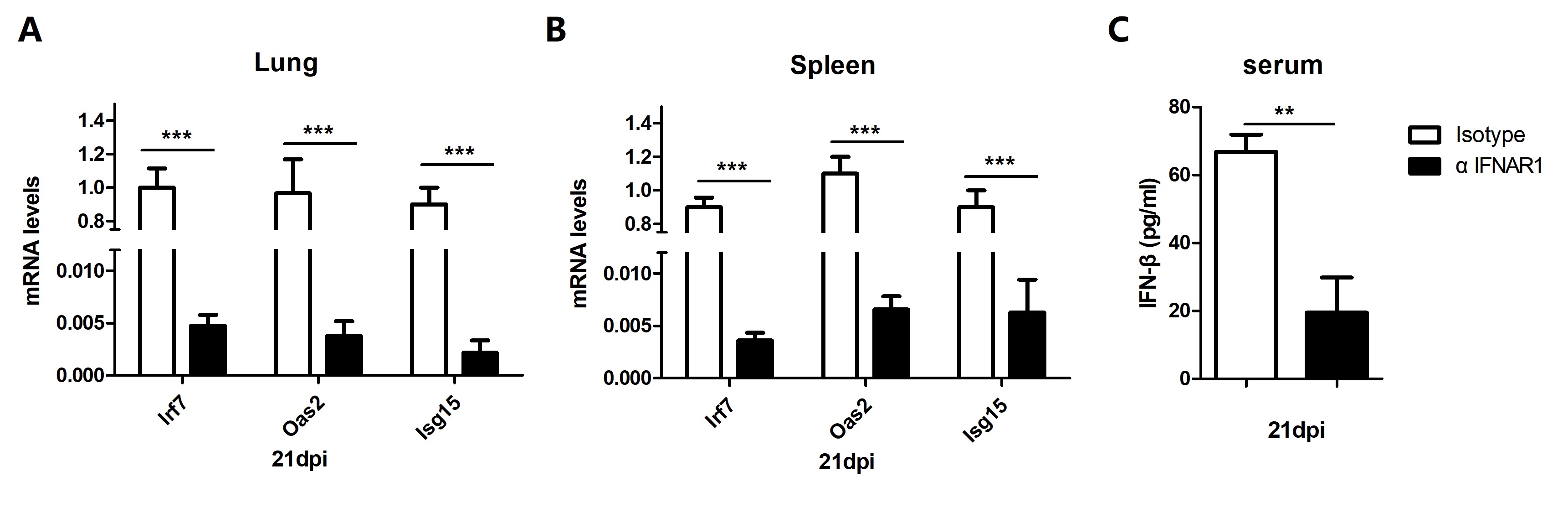
In each group, 12 mice C57BL/6 mice were treated twice with 500µg of anti-IFNAR1 antibody (clone MAR1-5A3; Biolegend San Diego, CA, USA) or mouse IgG1 (500µg) isotype control (clone MOPC21; Bio X Cell) one day prior to M. bovis infection and at 6 weeks post infection through intraperitoneal (i.p) injection. For the enumeration of total viable bacilli, lung and spleen tissues were lysed with small ceramic beads in phosphate buffered saline (PBS), in a tissue homogenizer apparatus (WKT technology) in accordance with the guidelines of manufacturer. An appropriate tenfold serial dilution was prepared in PBS. The dilutions were separately plated in triplicates on Middlebrook 7H11 agar supplemented with ampicillin (10 μg/ml) and sodium pyrovate (2-4mg/liter). After 2-3 weeks of incubation at 37°C, M. bovis colonies were counted [21].
IFNAR1 blocking antibody treatment in vitro
To investigate the inhibitory effect of Type I interferon signaling in vitro, BMDMs were treated with 1 µg/ml of IFNAR1 blocking antibody (Biolegend) for 2 hours. Then the cells were washed three times with warm PBS and infected with M. bovis at multiplicity of infection (MOI) 10 and incubated at 37oC for 24 hours.
IFN-β production assay
The level of IFN-β in the serum samples was determined by using ELISA assay as previously described (Cusabio, Wuhan, Hubei, China) according to the manufacturer’s protocols [21]. Briefly, 100 µl of standards and samples were added in respective wells of 96 wells antibody coated plate. After incubation and washing steps, conjugated secondary antibodies were added for 1 hour followed by same washing steps. Then substrate solution was added in each well followed by addition of stop solution. A standard curve was obtained by using 2-fold dilutions of the standard for each independent experiment. The concentration of cytokines was calculated using a standard curve.
Multiplex cytokine assays
The lung tissues were homogenized in PBS 0.05% v/v Tween 20 with a supplementation of protease inhibitor cocktail (Roche). The detection of multiple cytokines was carried out by using multiplex bead-based immunoassay kits (Millipore) according to the manufacturer’s protocol. MAGPIX® instrument (Luminex, USA) was used to perform multiplex bead-based immunoassay for the detection of multiple cytokines.
Quantitative RT-PCR assay
The cDNA synthesis from total RNA (50 ng) was performed by using the Revert Aid first-strand cDNA synthesis Kit (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s protocol. For the quantitative analysis of mRNA, real time-PCR (qRT-PCR) was performed by using AceQ qPCR SYBR Green Master Mix kit (Vazyme Biotech, Nanjing, China) according to the manufacturer’s instructions. β-actin was used as a house keeping gene for data analysis. The sequences of primers used in the current study are mentioned in Table 1. Amplifications were performed with the 700 Fast Real-Time PCR Systems (ViiA7 Real-time PCR, ABI). Thermal cycling conditions were 95°C for 5 minutes then 40 cycles at 95°C for 10 seconds and 60°C for 30 seconds. All fold changes were analyzed by using the ΔΔCt method [22]. Samples were measured in triplicate from three independent experiments.
Histopathology
For histopathology analysis, the left lung lobe was selected from mice of all experimental groups. 10 % formalin buffer was used as a tissue fixative solution and then the lung tissues were sectioned at 5 μm of thickness, followed by H&E or Ziehl-Neelsen staining methods. The inflammatory changes in the lung tissue was measured by observing the H&E stained lung sections under light microscopy at low (x10) and high (x40) magnifications. The superior lobes of the left lung were stained with H&E to assess the severity of inflammation; minimum 10 microscopic fields were selected for each section. The level of inflammation in the lung sections was quantified by measuring the area of lesion out of the total area of the section by using ImageJ software (National Institutes of Health, USA).
Immunohistochemistry
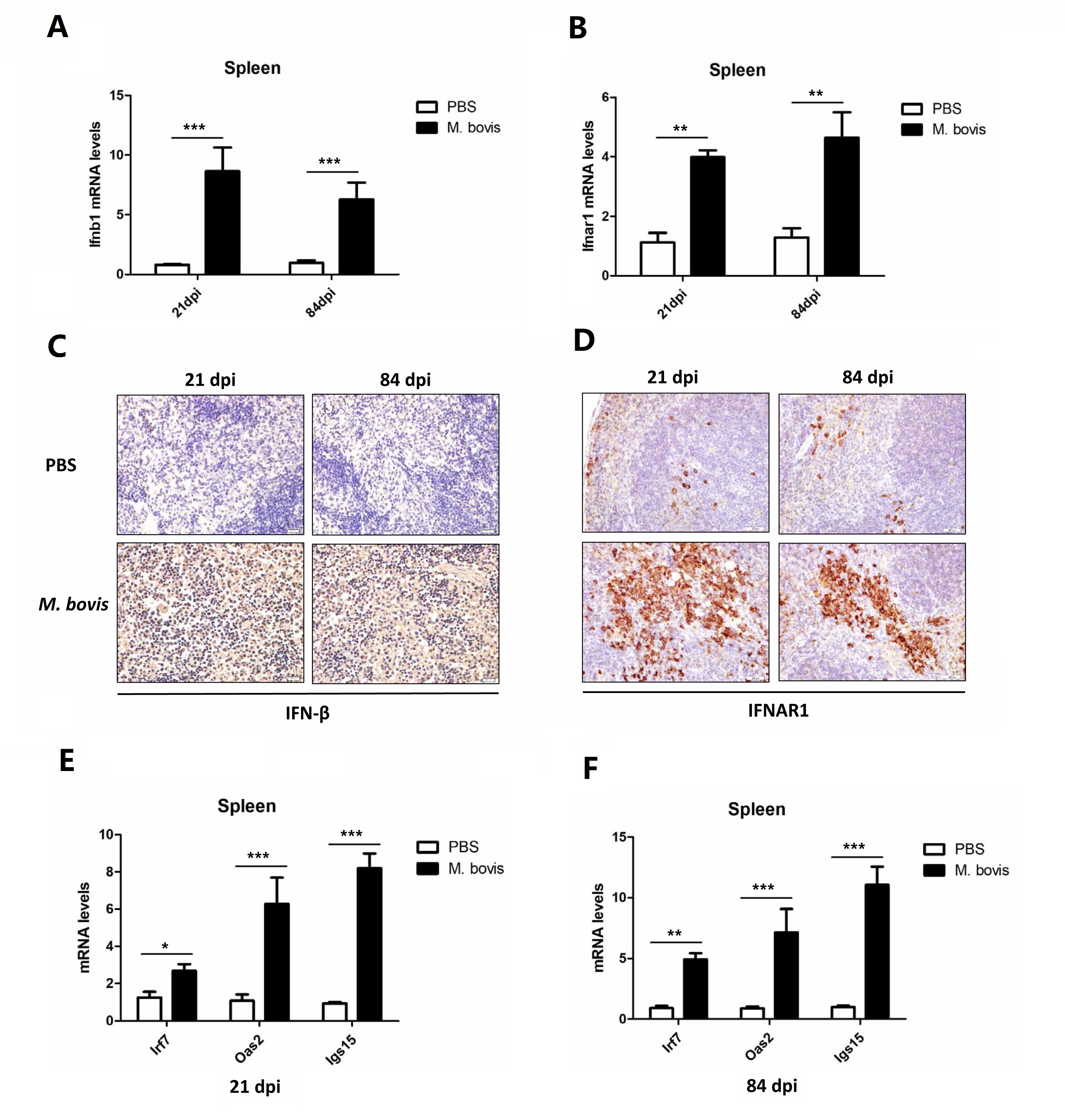
The lung tissues that were already fixed in formalin buffer (10%) were paraffinized and sectioned for immunohistochemical analysis. Briefly, after deparaffinization and antigen retrieval, the lung sections were blocked with BSA for 15 minutes at 37oC [26]. After blocking, the sections were incubated at 4oC with anti-mouse IFNAR1 (Biolegend) or anti-mouse IFN-β (Santa Cruz Biotechnology, CA, USA) overnight followed by HRP-labeled secondary rabbit anti-mouse IgG antibodies (Proteintech, Wuhan, China). The enzymatic activity was revealed by using 3, 3'-Diaminobenzidine (DAB). Digital images were collected on Olympus microscope fitted with DS-Ri2 camera. To quantify the intensity of IFNAR1 and IFN-β, 3 lung sections from independent animals of each group were visualized under low and high power of magnifications. The stained area compared with the total tissue area was determined by using Image-J software.
Lung cell isolation
Right lung lobes were washed with PBS before excision, and minced on the ice. The minced lung tissues were incubated with DMEM containing 50μl Collagenase 1 (1mg/m1) (Solarbio, Beijing, China) and 50μl DNase 1 (150U/m1) (Roche Biochemicals) for 1 hour at 37°C and shacked once every 15 minutes. After that tissue homogenates were passed through a cell strainer of 70μm pore size (BD Falcon, NY, USA) to make single cell suspension. Lung cells were centrifuged in complete DMEM for 5 minutes at 1000 rpm for obtaining leukocytes from cell suspension. After centrifugation, the supernatants were removed and the cell pellets were resuspended, and erythrocytes were lysed with Red Blood Cell Lysis Buffer (MultiSciences, Hangzhou, China) by keeping at room temperature for 5 minutes. About 10 ml of DMEM containing 10% FBS was added to stop the lysis reaction, and after centrifugation the supernatant was removed. After that the cells were resuspended in PBS and the number of cells was counted [26].
Flow cytometry
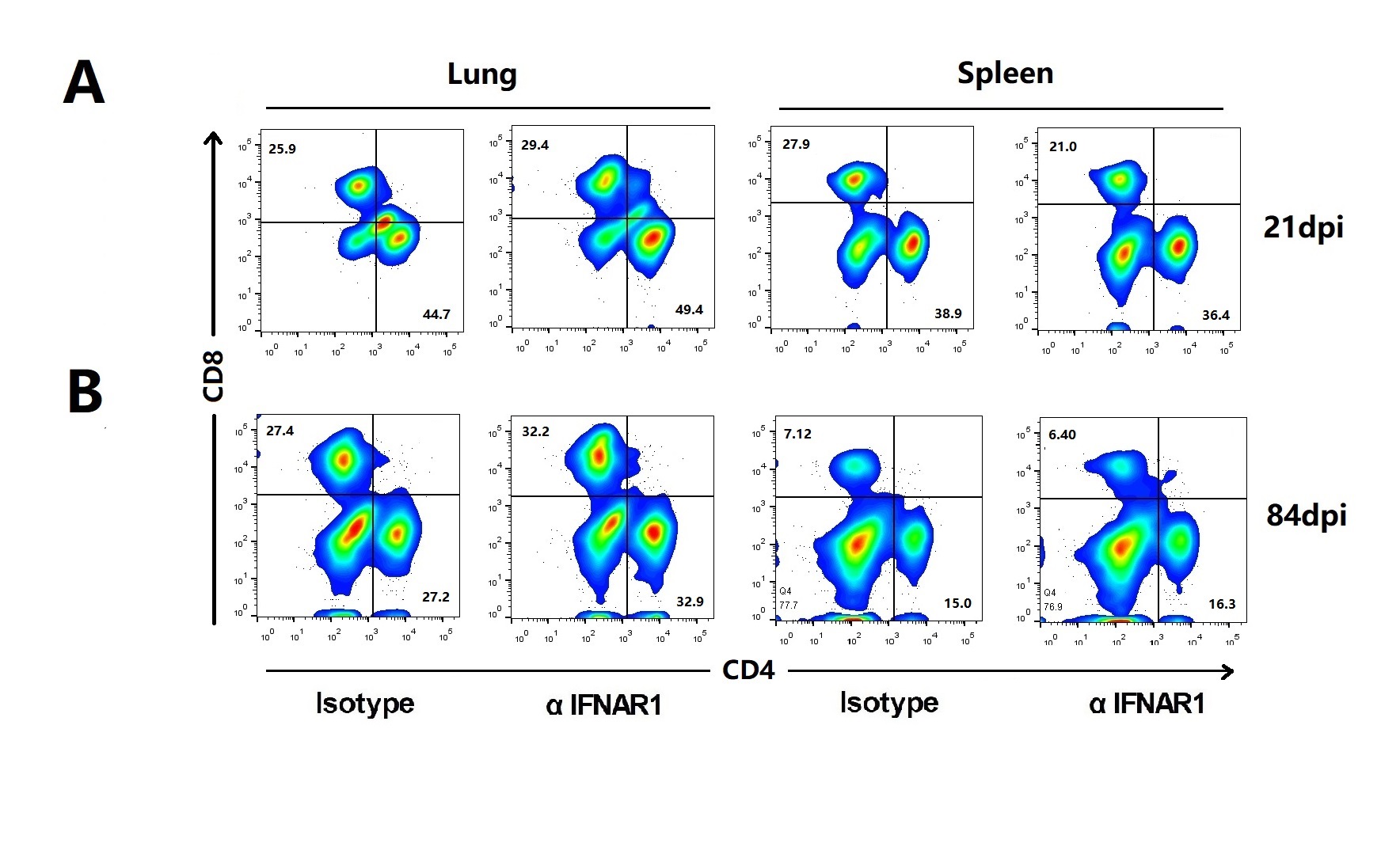
For differential quantification of innate immune cells, isolated leukocytes for both lung and spleen tissue were stained with antibodies against Ly6G PerCP-Cyanine5.5 (eBioscience, California, USA), CD11b-APC, CD11c-FITC, Ly6C-PE, CD11c-PE, MHC-II-FITC, CD40-FITC, CD80-FITC, CD86-FITC CD4-FITC and CD8-APC (all from eBioscience). For leukocytes analysis, single-cell suspensions from lungs were stimulated with 10 μg/ml of EAST-6 in the absence or presence of 4 μg/ml brefeldin A (Sigma, St. Louis, USA) for 6 h at 37°C [26]. CD206-PE (eBioscience) antibody was used for the detection of intracellular markers of lymphocyte subsets.
Statistical analysis
The data was analyzed by using GraphPad Prism 5 software USA. Student t test was applied for comparing between two groups. One way or two way ANOVA was applied for the comparison of multiple groups.
{kind=link}
{kind=link}
{kind=link}