Collections:
Specimens utilized for this analysis were opportunistically collected in Western North Carolina by a large volunteer network of State Park, National Park, and North Carolina Wildlife Resources Commission employees as well as amateur herpetologists. Permits were obtained from North Carolina Wildlife Resources Commission (#21-ES00592), Blue Ridge Parkway (#BLRI-2021-SCI-0009), and North Carolina State Parks (#2021_0520). Collections occurred from 2019–2022 and included scale clips, muscle, liver, or shed skin tissues from nuisance animals within state parks, animals found dead due to road mortality, or animals in gestation sites. All tissues, except shed skins, were stored in 95% ethanol at -20°C until DNA extraction. Location information was recorded with each collection, and GPS coordinates were later extracted from Google Maps.
DNA isolation:
DNA was extracted with a PureLink Genomic DNA mini-kit (Invitrogen, Carlsbad, California, USA) following the manufacturers protocol with minor modifications. Resulting DNA was examined for quality with a Nano-Drop 1000 (Thermo Fisher Scientific, Waltham, MA, USA) and a 1% agarose gel. High quality DNA samples were diluted to 30 ng/µl and randomly arrayed into 96 well plates with both positive and negative (ddH2O) controls to ensure the accuracy of genotyping.
Microsatellite analysis:
A total of 35 previously published microsatellites markers were screened against a small number of individuals to identify polymorphic loci that easily amplified (Villarreal et al., 1996, Holycross et al., 2002, Pozarowski et al., 2012). These 35 primer pairs were modified to include an M13 tag (5’-CACGACGTTGTAAAACGAC-3’) on the 5’ end of the forward primer to facilitate fluorescent tagging of PCR products with FAM, VIC, NED, or PET fluorophores (Life Technologies, Grand Island, NY, USA) (Schuelke, 2000). A total of 19 markers with diverse repeat motifs were utilized to genotype samples including 7–87, CW_A14, CW_A29, CW_B23, Ca120, Ca122, Ca131, Ca139, Ca227, Ca238, Ca264, Ca274, CS2322, CS234, CS2340, CC1110, CC2210, CC227, CC2333 (Table 1). PCR reactions were prepared in 10µL volumes consisting of GoTaq Flexi Buffer, 2.5 mM MgCl2, 800 µM dNTPs, 0.5 µM of reverse primer, 0.25 µM of tagged forward primer, 0.25 µM of a M13 fluorescent labeled primer, 0.5 units of GoTaq Flexi DNA Polymerase, and ~ 30ng of DNA (Promega, Madison, Wisconsin, USA). A touchdown PCR protocol was employed to increase specificity and reduce stutter on an Eppendorf Mastercycler thermal cycler (Eppendorf, Hauppauge, NY, USA) (Korbie and Mattick, 2008). Uniquely tagged PCR products were then multiplexed with HI-DI and a GeneScan Liz 500 size standard (Applied Biosystems, Foster City, California, USA) for separation on an ABI 3730. The chromatograms were scored in Geneious Prime 2023.2.1 using the microsatellite plug-in (Biomatters, Auckland, NZ) (Kearse et al., 2012).
Table 1
Microsatellite markers utilized in this study. Including original marker name, publication, Fluorophore utilized, the observed size range and microsatellite repeat motif.
| Marker name | Publication | Fluorophore | Observed product sizes (bp) | Repeat motif |
| 7–87 | Villarreal et al., 1996 | NED or PET | 161–181 | CA |
| CW_A14 | Holycross et al., 2002 | NED | 153–187 | AC |
| CW_A29 | Holycross et al., 2002 | VIC | 166–190 | AC |
| CW_B32 | Holycross et al., 2002 | PET | 220–266 | TG_AG |
| Ca120 | Pozarowski et al., 2012 | FAM | 270–320 | AG |
| Ca122 | Pozarowski et al., 2012 | VIC | 187–213 | TG |
| Ca131 | Pozarowski et al., 2012 | NED | 227–263 | TCT |
| Ca139 | Pozarowski et al., 2012 | PET | 335–395 | TTA(TTG)2(TTA)2 |
| Ca227 | Pozarowski et al., 2012 | VIC | 300–486 | TCC |
| Ca238 | Pozarowski et al., 2012 | NED | 300–360 | AGGC |
| Ca264 | Pozarowski et al., 2012 | PET | 325–413 | AAAG |
| Ca274 | Pozarowski et al., 2012 | FAM | 294–300 | GT |
| CS2322 | Pozarowski et al., 2012 | VIC | 431–559 | GATA |
| CS234 | Pozarowski et al., 2012 | NED | 424–512 | GAAA |
| CS2340 | Pozarowski et al., 2012 | PET | 256–278 | TG - Complex |
| CC1110 | Pozarowski et al., 2012 | FAM | 270–326 | CTTT - Complex |
| CC2210 | Pozarowski et al., 2012 | VIC | 314–358 | GAAA |
| CC227 | Pozarowski et al., 2012 | NED | 221–293 | CTTT |
| CC2333 | Pozarowski et al., 2012 | PET | 423–483 | CTA |
Management unit determination analysis:
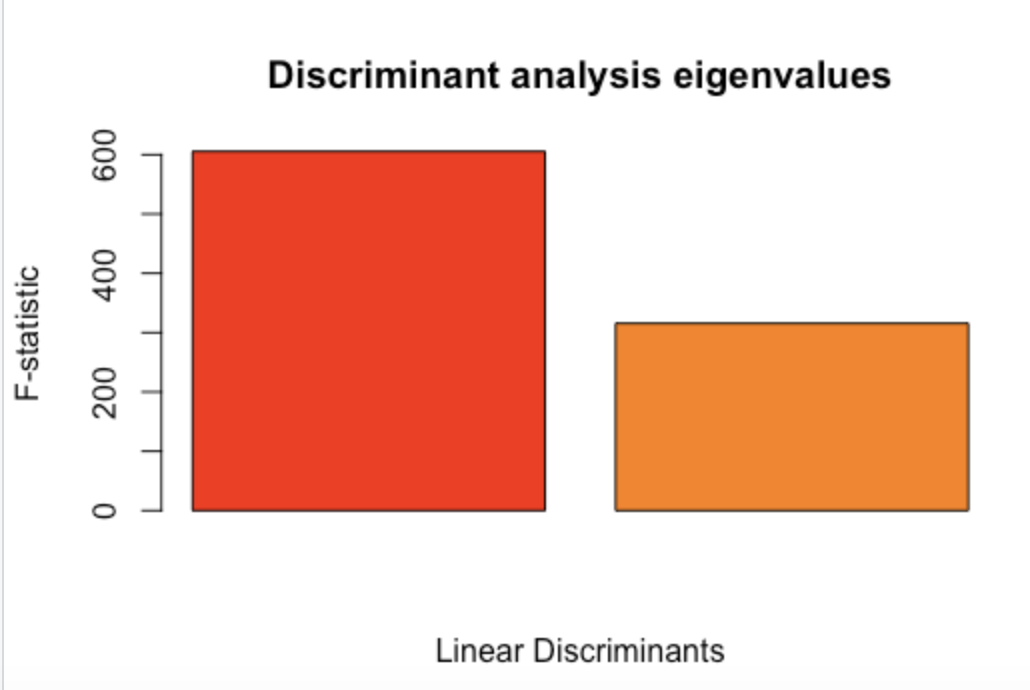
The study’s sampling strategy utilized did not allow for a priori determination of the population of origin for different samples. Therefore, we investigated population structure using a discriminant analysis of principal coordinates (DAPC) and STRUCTURE analysis with the R package poppr and STRUCTURE software (Kamvar et al., 2014; Pritchard et al., 2000).
The STRUCTURE analysis began by searching for the optimal number of clusters (K). We tested initial K values ranging from 1 to 10, with burn-in periods set to 100,000 and MCMC repetitions set to 500,000 (Pritchard et al., 2000). The resulting values were examined using the pophelperShiny app to identify the optimal K value utilizing the Evanno method (Evanno et al., 2005; Francis, 2017).
Once the optimal K was identified, STRUCTURE was rerun using this K value, with burn-in periods set to 500,000 and MCMC repetitions set to 1,000,000. Samples were then grouped into management units based on the Q-matrix score. Georeferenced points for each sample were mapped using ArcGIS Pro, and an Inverse Distance Weighted (IDW) interpolation model was employed through the Spatial Analyst Toolbox to predict the distribution of management units across the collection area. (ArcGIS Pro, 2023).
Genetic diversity estimates:
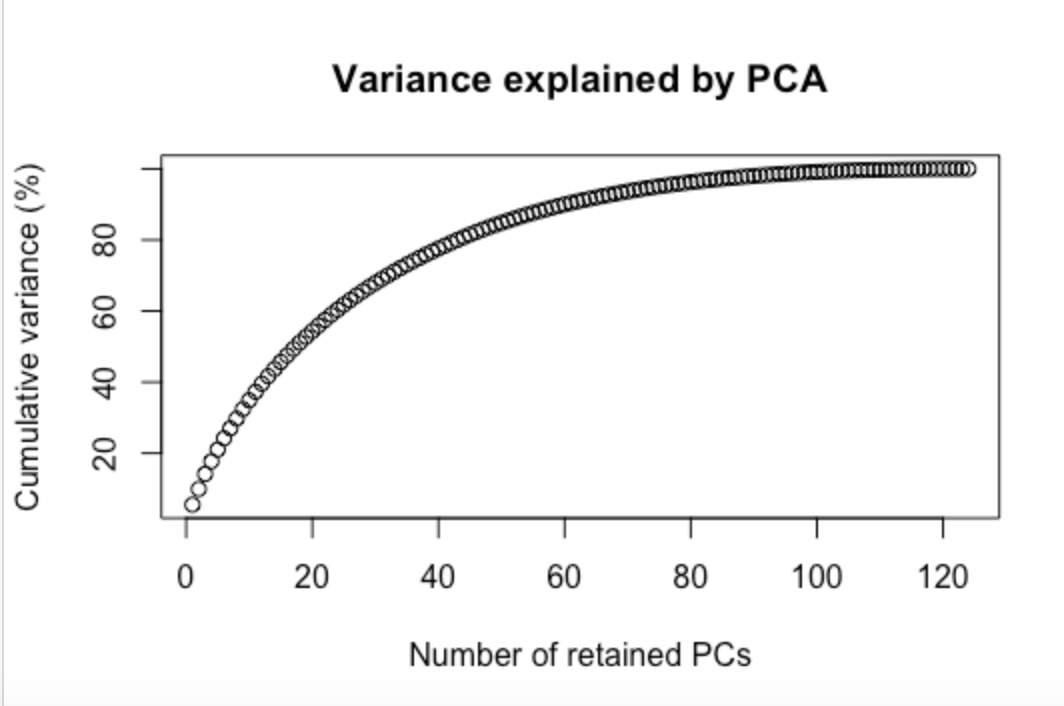
The resulting genotype data were analyzed using the R package poppr and the Excel-based package GenAlEx 6.51b2 (Peakall and Smouse, 2012; Kamvar et al., 2014; R Core Team, 2021). A genotype accumulation curve was generated to verify the power of the markers in distinguishing unique individuals. Subsequently, we calculated several genetic diversity metrics by locus: the number of alleles observed, Simpson's Index of Diversity, expected heterozygosity, and evenness.
Finally, using management unit designations from the STRUCTURE analysis Q-matrix score, we calculated: the number of multi-locus genotypes, mean number of alleles per locus, mean number of effective alleles per locus, observed and expected heterozygosity, Shannon-Weiner Diversity Index, Simpson's Index, and evenness for each group. Classic measures of population differentiation (FST, GST) and estimates of the number of migrants were also calculated.
Genetic bottleneck test
We further examined the Q-matrix score management unit designations for signs of a recent genetic bottleneck. To do this, we used BOTTLENECK version 1.2.02 (Piry et al., 1999) to test for deviations from the mutation-drift equilibrium model. The analysis was conducted using two different mutational models: the stepwise mutation model (SMM) and the two-phase model (TPM). We employed a sign test, a standardized differences test and a Wilcoxon signed-rank test to assess significance under both models. Additionally, we examined allele frequency distributions to identify the L-shaped curve expected under the mutation-drift equilibrium model (Luikart et al., 1998).
{kind=link}
{kind=link}