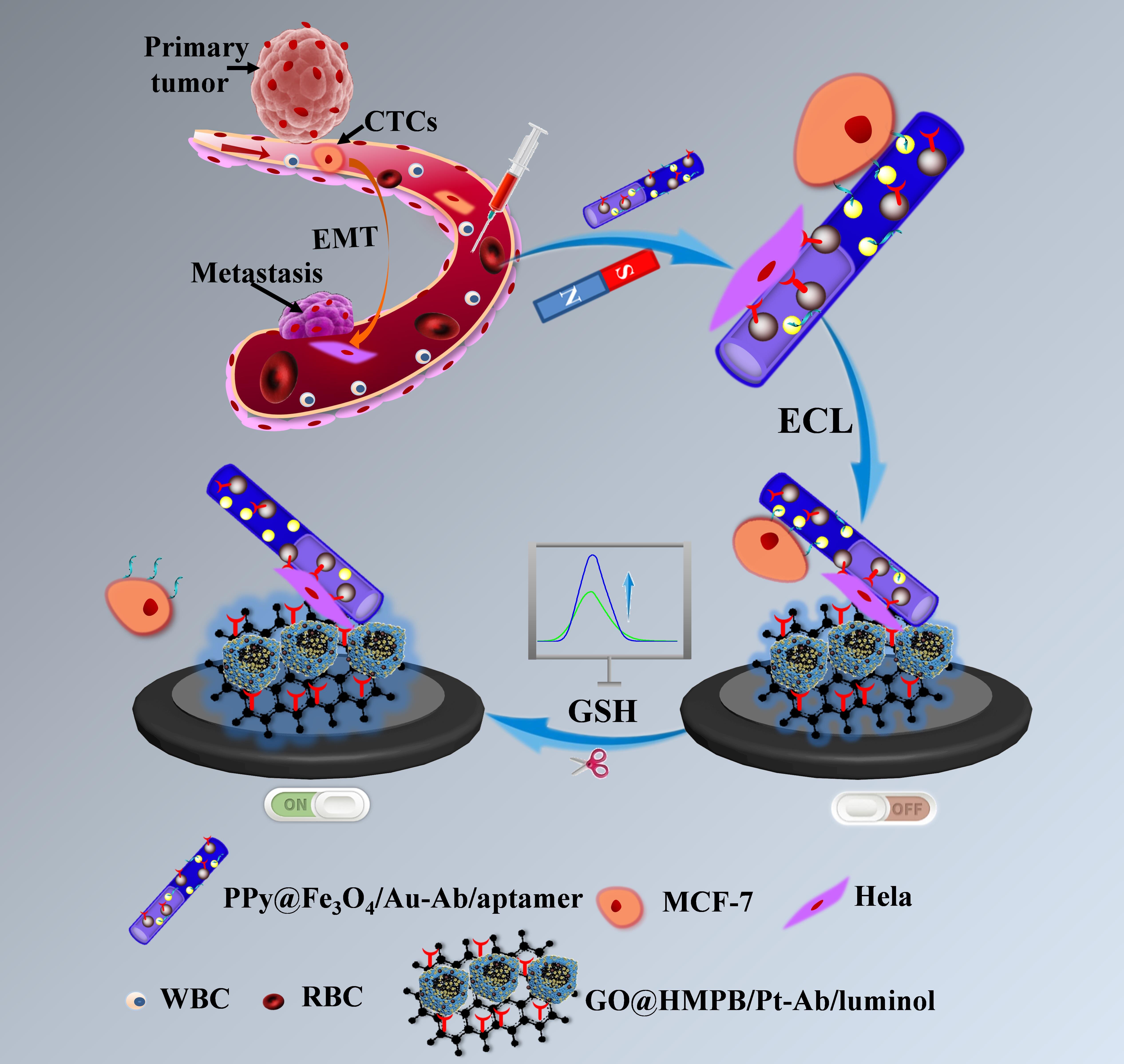
Principle for Phenotypic Enumeration of CTCs in Blood
The designed strategy of this ECL cytosensor is illustrated in Scheme 1. Initially, PPy@Fe3O4/Au nanotubes were synthesized via a one-pot method using MoO3 nanorods as templates. During this process, the polymerization of pyrrole, formation of Fe3O4 as well as Au NPs and removal of MoO3 templates occurred simultaneously. Generally, the EpCAM and N-cadherin are known to be highly expressed on MCF-7 (epithelial type of CTCs) and Hela cells (mesenchymal type of CTCs), respectively. Moreover, the SYL3C and NC3S aptamers have been reported to exhibit specific recognition towards epithelial and mesenchymal CTCs. Subsequently, the antibody/aptamer combination of anti-EpCAM/NC3S and anti-N-cadherin/SYL3C were immobilized onto PPy@Fe3O4/Au due to the abundant carboxyl groups of Fe3O4 and Au-S bond (Scheme 1A). In addition, GO@HMPB/Pt-luminol signal probe was obtained as shown in Scheme 1B. The two-dimensional GO, possessing excellent conductivity and large specific surface area, served as a carrier for anti-EpCAM and HMPB/Pt NPs loaded with substantial amounts of luminol. HMPB exhibits a hollow structure and possesses a significant quantity of mesoporous sites suitable for loading Pt NPs and luminol. Scheme 1C depicts the diagram for capturing, magnetically separating and electrochemical phenotypic counting two types of CTCs in whole blood with the nanozymes-catalyzed cascade amplification. Upon incubation with a blood sample, the PPy@Fe3O4/Au dual-target capture probe effectively recognizes and captures target MCF-7 and Hela cells. The external magnet facilitates the selective separation of target cells from a large excess of other cells, such as leukocytes and erythrocytes. GO@HMPB/Pt-luminol signal probe conjugated with EpCAM antibody was modified onto the glassy carbon electrode to bind with the enriched CTCs and report the corresponding ECL intensity. Peroxidase-like HMPB and high activity Pt NPs synergistically catalyze the H2O2 into reactive oxygen species and enhance the ECL intensity significantly. However, the ECL intensity was attenuated upon the capture of CTCs onto electrode and subsequently restored by cleaving the Au-S bond through GSH to selectively release epithelial or interstitial CTCs.
Characterization of the PPy@Fe 3 O 4 /Au and GO@HMPB/Pt Nanocomposites.
The micromorphology and elemental composition of the synthesized nanomaterials were characterized by SEM and TEM firstly. PPy@Fe3O4/Au has been synthesized and its magnetic as well as catalytic properties were preliminarily studied in our previous work[21]. The detailed structure and element composition were further studied with TEM and energy dispersive X-ray spectroscopy (EDS) elemental mapping, as shown in Fig. 1A and Figure S1. TEM images showed a typical tube-like structure uniformly loaded with Fe3O4 and Au NPs. The magnified image (insert) indicated slight agglomeration of the Fe3O4 and Au NPs due to the anisotropic dipolar interactions. Furthermore, the mapping images corresponding to Fe, Au, C, N and O elements provided additional evidence for the successful formation of PPy@Fe3O4/Au.
The TEM image of GO synthesized using the electrochemical exfoliation exhibited a thin lamellar morphology, in accordance with previous literature reports (Fig. 1B). The SEM and TEM images (Fig. 1C, 1D) clearly showed the cubic structure of PB NPs converted to distinct hollow structure of HMPB after etched by hydrochloric acid. The TEM image (Fig. 1E, Figure S2) confirmed that the HMPB and Pt NPs were successfully supported on GO. As shown in Fig. 1F-J and L, high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and the corresponding elemental mapping were conducted to further confirm the distribution of HMPB and Pt NPs on GO. The high-resolution transmission electron microscopy (HRTEM) image (Fig. 1K) demonstrated that GO@HMPB/Pt possessed a lattice fringe spacing of 0.226 nm, corresponding to the (111) plane of metallic Pt NPs. These above results confirmed the successful preparation of the GO@HMPB/Pt. Meanwhile, the morphology and elemental distribution of the GO@PB/Pt composite were characterized. As depicted in Figure S2, PB exhibited a solid cubic particle while Pt nanoparticles were uniformly dispersed on both the surface of PB and GO.
SERS and FTIR were adopted to characterize the functional groups on GO@ HMPB/Pt. From the SERS spectra (Fig. 2A), the typical D-band (1352 cm− 1) and G-band (1591 cm− 1) peaks for GO and the characteristic peak for C ≡ N band at 2163 cm− 1 of HMPB were observed. The D-band/G-band intensity ratio of GO was significantly lower compared to Hummers' method, indicating that the mild oxidation process results in a reduced presence of D-defects [22]. FTIR spectra (Fig. 2B) provide insights into characteristic functional groups including -OH (3273 cm− 1), C = O (1641 cm− 1), C-O (1545, 1373, 1045 cm− 1) and C ≡ N (2079 cm− 1). Subsequently, the crystal structure of GO@HMPB/Pt was confirmed by XRD in Fig. 2C. The diffraction peaks at 17.4°, 25°, 35°, 39.5°, 50.6°, 54°, 57.3°, 66° and 68° are associated with PB (JCPDS NO.73–0687) [23]. The sharp peak at 26.5° of GO [24] and two weak peaks at 40.1°and 46.2°correspond to Pt NPs (JCPDS NO. 04-0802) [25]were emerged after combination. The XPS spectrum show typical peaks of Pt 4f, C1s, N1s, O1s and Fe2p from 0 to 1000 eV (Figure S3). Figure 2D shows the N 1s spectrum, with 397.04, 398.38, 400.3, and 402.6 eV correspond to C ≡ N, C-N and N-H. The Fe2p spectrum exhibits two contribution, Fe 2p3/2 and Fe 2p1/2, and four binding energy peaks were located at 707.8, 712.7, 721.4 and 724.9 eV, which can be assigned to the existence of [Fe(CN)6]4− and Fe3+ (Fig. 2E). The detailed spectrum of C1s and Pt4f were shown in Figure S3. All the peak positions confirm the presence of predominantly surface end groups such as -COOH, -OH, and –NH2 on the GO@HMPB/Pt surface, providing extensive sites for antibody loading. The Brunauer–Emmett–Teller (BET) analysis in Fig. 2F reveals that the surface area of HMPB is approximately 110.4 m2/g, which is larger than PB (49.5 m2/g).
Construction of the ECL Cytosensor and Feasibility Tests
After confirming the successful synthesis of the capture vector and the signal probe, the ECL cytosensor was constructed. As displayed in Fig. 3A, rapid separation of PPy@Fe3O4/Au was achieved within 7 s using a magnet, which demonstrated good magnetic properties of PPy@Fe3O4/Au. Additionally, the changes observed in Figure S4 regarding the Zeta potential demonstrate the successful fabrication of the PPy@Fe3O4/Au dual-target magnetic probe and its proficient capability to capture CTCs. A standard chromogenic system consisting of TMB and H2O2 was employed to investigate the peroxidase-like of GO@HMPB/Pt. As shown in Fig. 3B, a typical blue color appears in the GO@HMPB/Pt + TMB + H2O2 system, while it appears nearly colorless in control experiments without H2O2, TMB or GO@HMPB/Pt. The corresponding UV-vis absorbance spectra of GO@HMPB/Pt + TMB + H2O2 system show a prominent peak at 625 nm, which confirms the inherent peroxidase-like activity of GO@HMPB/Pt. Additionally, the superior performance of GO@HMPB/Pt compared to PB, HMPB and HMPB/Pt is verified by the deepest color and strongest absorbance at 625 nm generated due to GO@HMPB/Pt (Figure S5). Additionally, the zeta potential gradually becomes negatively charged, indicating the successful assembly of the cascade amplifying signal molecule (Figure S6). The peroxidase mimics of GO@HMPB/Pt serve not only as a redox mediator to H2O2 but also as a scaffold carrier to immobilize luminol to form a solid-state luminophore, which accelerates the generation of reactive oxygen species and decreases the distance between the luminophore and electrode. The loading capacity of HMPB/Pt was determined through UV-vis absorption spectroscopy and the loading capacity of luminol by HMPB/Pt was calculated to be as high as 91.5% (Figure S7).
The technique of electrochemical impedance spectroscopy (EIS) offers comprehensive insights into the modification process of the ECL cytosensor. As illustrated in Fig. 3C, the progressive increase in electron-transfer resistance (Ret) indicates the successful modification of the glassy carbon electrode step-by-step. Especially, the Ret values increased significantly after CTCs captured on GCE. However, when the Au-S bond was cleaved by GSH to release Hela cells, decrease of Ret value was observed. This result indicates that the ECL cytosensor was feasible for simultaneous phenotypic typing and quantification of CTCs. Meanwhile, the universality of prepared cytosensor to epithelial or interstitial cell release was further verified by ECL response performance while adjusting the type of recognition probe. Figure 3D displays the histogram of ECL response during the construction of CTCs cytosensor. The peroxidase-like activity of GO@HMPB/Pt acts as a co-reaction accelerator to cascade amplify signal of luminol (column a). Due to the high impedance of biomolecules, the ECL intensity gradually decreases with the introduction of double-target probes (column b), interstitial Hela (column c) and mixed Hela and MCF-7 (column d). The cleavage of the Au-S bond by GSH resulted in the release of interstitial Hela cells and partial recovery of ECL response, which was consistent with the result of EIS. Meanwhile, the ECL sensor achieved comparable results by replacing the recognition molecules of the double-target magnetic probe with N-Cadherin and SYL3C to release epithelial MCF-7 cells (Figure S8). These above results demonstrate the successful construction of the ECL immunosensor, which possesses a universal capability to capture and release CTCs exhibiting diverse phenotypes.
Phenotypic Counting of CTCs in PBS Buffer
To evaluate the analytical performance of the ECL cytosensor for phenotypic enumeration of CTCs, samples containing different concentrations of Hela, MCF-7 and a mixture of dual cells (1:1) in PBS buffer were analyzed by the ECL sensor. Since the ECL cytosensor was constructed based on specific recognition and electron transfer rates on electrode surface, the concentration of carrier, recognition probe, luminol, GSH and pH value of the electrolyte were optimized prior to investigating the analytical performance of the cytosensor. As seen in Figure S9, the efficient isolation of CTCs can be achieved using 1 mg/mL of PPy@Fe3O4/Au. The carrier of GO (0.2 mg/mL), recognition probe of anti-EpCAM (0.5 µg/mL), N-Cadherin antibody (0.5 µg/mL), NC3S (5 µM), luminophor of luminol (0.5 mM), GSH (5 µM) for cleaving Au-S bond and pH = 8 were optimized and selected for the following electrochemical tests. Then, the performance of the ECL sensor on CTCs with a single phenotype was validated. The ECL intensity gradually decreased as the concentration of Hela or MCF-7 cells increased as the capture of cells onto electrode inhibited electron transfer rates at the electrode surface (Fig. 4A, C). The relationship between the peak intensity and the logarithm of Hela or MCF-7 cell concentrations showed linearity within the ranges from 1 to 10000 cells/mL and 1 to 5000 cells/mL, with R2 values of 0.995 and 0.999, respectively (Fig. 4B, D). These results validate that the constructed ECL sensor is universally applicable for both enrichment and detection of single-phenotype CTCs. Then, to demonstrate the cell phenotypic counting capacity of ECL sensor, we performed simultaneous detection of Hela and MCF-7 cells as epithelial and mesenchymal models to analyze the EMT phenotype of tumors. As illustrated in Fig. 4E, the ECL intensity exhibited a negative correlation with cell concentration, but showed recovery following cleavage of Au-S bond by GSH. Two distinct linear segments were independently acquired with linear correlation coefficient of 0.993 and 0.998. On the basis of S/N = 3, the calculated limit of detection (LOD) for mixed and epithelial were 2 cells/mL and 1 cells/mL, respectively. Compared to previously reported methods, the ECL sensor presented here not only enables simultaneous detection of multi-phenotype CTCs, but also provides accurate quantification of single-phenotype CTCs with higher sensitivity and lower LOD (Table 1).
Table 1
Comparison of the analytical performance for CTCs detection.
| Technique | Target cell | Linear range (cells mL− 1) | Limit of Detection (cells mL− 1) | reference |
| Fluorescence | A549 MCF-7 | 5–5×103 5–5×103 | 2 4 | [26] |
| EIS | MCF-7 | 1–40 | 3 | [27] |
| Square Wave Voltammetry | MCF-7 | 5–5×105 | 5 | [28] |
| Amperometry | MCF-7 | 10 − 1×104 | 3 | [29] |
| ECL | MCF-7 | 10 − 1×104 | 8.91 | [30] |
| ECL | MCF-7 | 8 − 1×105 | 2 | [31] |
| Photoelectrochemistry | MCF-7 | 50 − 1×107 | 18 | [32] |
| ECL | MCF-7 Hela | 1–5×103 1–1×104 | 2.48 and 1.8 | This work |
To investigate the specificity of the established ECL sensor, a range of control compounds including ions (Zn2+, Na+, K+, Mg2+, I−, Cu2+, SO42−, Cl−) at a concentration of 2 mM, molecules (UA, AA, DA, GSH, glucose) at a concentration of 10 mM, proteins (BSA, Protein S, Arg, Gly, Recombinant, MUC1) at a concentration of 5 µM and other cells (HL-7702, DU145, BEAS-2B, Hepg2) at a concentration of 10000 cells/mL were analyzed using the designed sensor (Fig. 5A). Despite the abundance of other compounds in comparison to the blank experiment, no significant decrease in peak intensity was obtained. Additionally, the coexistence of target CTCs with other compounds does not alter the ECL responses, confirming the exceptional interference resistance capability of this sensor. Furthermore, the cycling and long-term stability of ECL sensor were studied. As shown in Fig. 5B, the relative standard deviations (RSDs) for the ECL response of target cells (Hela/MCF-7 = 1:1 with a total 1000 cells/mL) were found to below 3% undergoing 12 cycles. Additionally, the ECL intensity exhibited minimal decline even after a duration of 12 days, indicating good stability of the cytosensor (Figure S10A). Moreover, the reproducibility of the ECL immunosensor was evaluated by preparing six independent electrodes for the determination of 2000 cells/mL target CTCs, and the RSD was calculated to be 0.507% (Figure S10B). In summary, the above results further validate the reliability of the proposed ECL platform for precise phenotypic counting of CTCs.
Phenotypic Enumeration of CTCs in Whole Blood
The capture and phenotypic enumeration of CTCs in human whole blood were further researched to explore the potential clinical applications of the ECL sensor. Different concentrations of CTCs with the ratio of Hela to MCF-7 as 1:1 were spiked into the whole blood that obtained from a healthy volunteer without any pretreatment and the CTCs were then separated by the magnetic PPy@Fe3O4/Au dual-target probe. Then, the corresponding ECL response obtained due to the capture and the release of CTCs was recorded for phenotypic enumeration analysis. The analysis results of CTCs in whole blood and PBS agreed well for detection of two types of CTCs (Fig. 6A) and single phenotypic CTCs (Fig. 6B). Although the ECL responses in whole blood exhibit a slight decrease compared to that in PBS buffer at low concentration, the accurate phenotypic enumeration of CTCs can still be effectively achieved, which suggest that our developed ECL sensor exhibits significant potential for application in the precise detection of CTCs in whole blood for personalized diagnosis of cancer.
{kind=link}