3.1. Preparation and identification of TDH
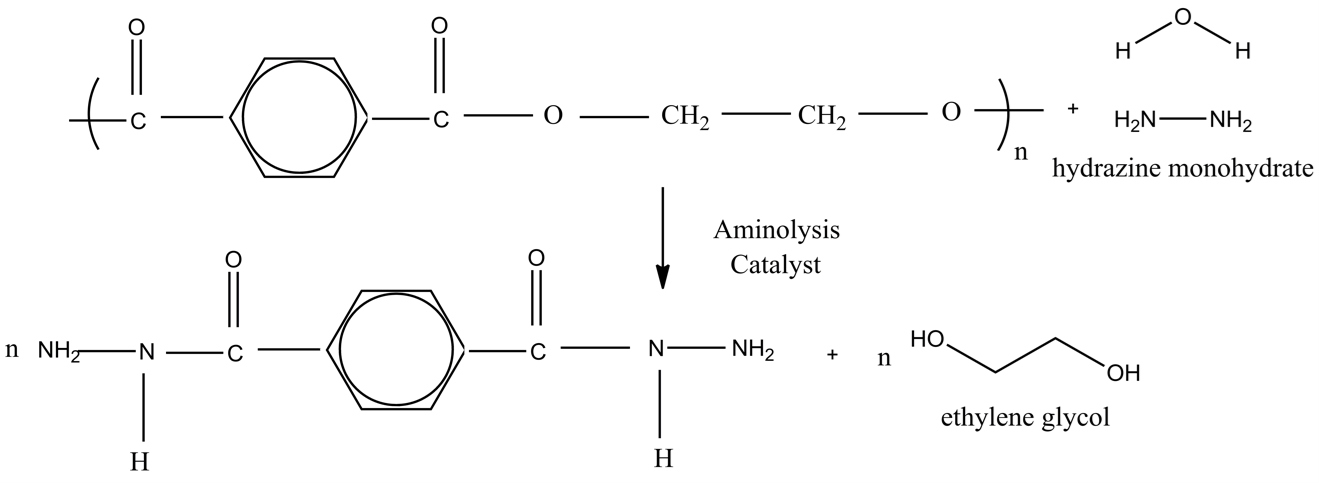
The aminolysis of PET was conducted using HMH as the amine reagent, catalysts, and DMSO, involving PET flakes sourced from recycled post-consumer bottles, as depicted in Scheme 1.
FTIR spectrometry was used to analyze and confirm the structure of TDH derived from the aminolysis of PET. The spectrum depicted in Fig. 1 displays a carbonyl absorption peak, identified as a primary amide, at 1633 cm–1. An observed peak at 1545 cm–1 corresponds to the bending vibrations of NH2 or NH (secondary amide bond). A broad and sharp peak at 3327 cm–1 is attributed to the N–H stretching vibration; its absence in the PET spectrum confirms the degradation of the polymer and the formation of amide groups. The peak at 1340 cm–1 results from the C–N stretching vibration, indicating the breaking of the C–O bond in PET and subsequent bonding of the carbon to the nitrogen atom in HMH. Additionally, aromatic C = C and C–H stretching vibrations are noted at 1336 cm–1 and 3050 cm–1, respectively. Sharp peaks at 1490 cm–1 and 1541 cm–1 are associated with the presence of the aromatic ring, observable in both the synthesized TDH and PET. These results from FTIR spectroscopy confirm that the degradation of PET and the production of TDH as a depolymerization product have been successfully achieved.
Figure 2 displays the 1H NMR spectrum of the product obtained from the aminolysis of PET flakes. This spectrum features peaks at 7.89 ppm (singlet, 4H, aromatic protons), 9.9 ppm (singlet, 2H, NH from –CONHNH2), and a broad peak at 4.58 ppm (singlet, 4H, NH2 from –CONHNH2). The spectral data for the aminolyzed product from PET flakes align well with the values reported in the literature for TDH.[22–24] The 1H NMR spectrum complements the findings from the FTIR analysis, providing a cohesive understanding of the product's structure.
The differential scanning calorimetry (DSC) thermograms, displayed in Fig. 3, compare the thermal behavior of a sample obtained from PET depolymerization and PET flakes across two continuous thermal scans. The thermogram in Fig. 3 reveals the start of an endothermic peak at 328°C with a narrow and elongated distribution, indicative of the melting point of the synthesized product. This peak contrasts with the shorter and broader peak of the PET flakes, aligning with the reported melting point of terephthalic dihydrazide in the literature. The melting point of terephthalic dihydrazide has previously been reported by Shukla and George to fall within this range.[25, 26] The DSC results confirm the formation of terephthalic dihydrazide as the final product of the aminolysis of PET flakes in this study. The presence of an aromatic ring in the structure of terephthalic dihydrazide contributes to its high thermal stability and delays its decomposition.
An exothermic peak at 336°C signifies the total decomposition of the product at that temperature, highlighting a close temperature range between melting and TDH degradation. Meanwhile, the melting peak for the utilized PET sample appears at approximately 245°C, showing a significantly wider temperature gap between the melting and degradation points compared to the small TDH molecule.
According to Fig. 4, thermogravimetric analysis curves for the TDH sample and PET flakes are presented side by side for comparison. The TGA diagram, shows TDH remains stable up to 300°C, and the initial weight loss (d5%) of the synthesized sample occurs at a temperature above 300°C. This loss could be due to residual moisture or small molecules resulting from depolymerization despite the sample being dried in an oven at 60°C for 24 h. In contrast, the initial weight loss for PET flakes starts at around 380°C. Additionally, the TGA, DTA, and DTG curves shown in Fig. 5 highlight three major areas of weight change in the synthesized TDH. The behavior of the TGA curve and the peaks observed in the DTG and DTA curves indicate that the melting temperature of TDH, resulting from depolymerization, is 328 degrees Celsius. This finding is consistent with the results reported by other researchers. Furthermore, this temperature obtained from the thermal gravimetric analysis aligns perfectly with the results from the differential scanning calorimetry tests.[25, 26]
3.3. Preparation and identification of oligomer and copolyurea
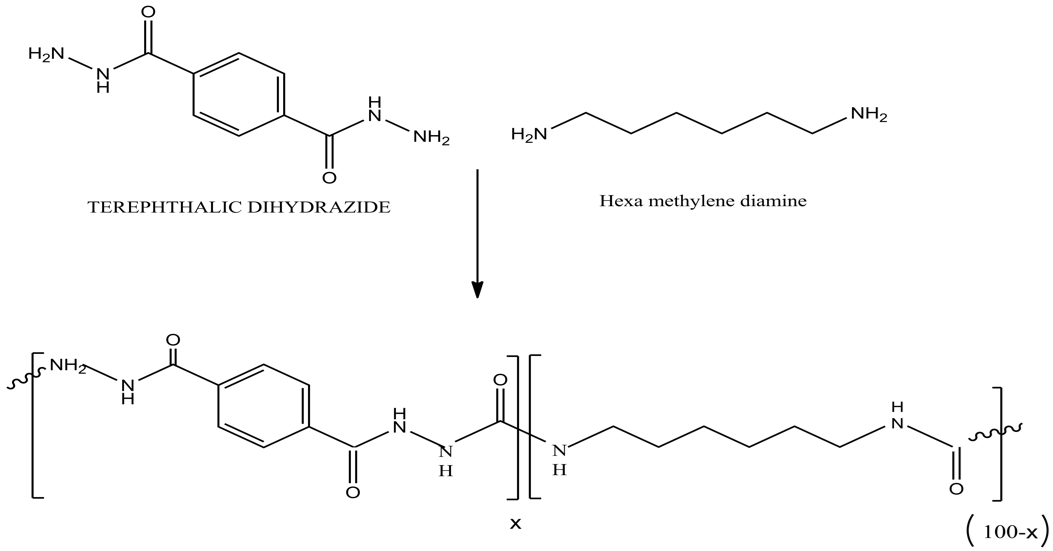
Polyureas were produced using a two-step polymerization process involving diamines and CO2, as outlined in Scheme 2. As previously discussed, the synthesis process comprises an initial stage of oligomer preparation, followed by the creation of copolymer samples. To achieve the best conditions for the synthesis reaction, various tests were performed based on the materials’ structure and findings from other research studies. Consequently, to identify the appropriate conditions for synthesizing HDA and TDH-containing copolyurea in a high-pressure autoclave reactor, the optimization of the initial stage of synthesis, known as oligomerization, has been conducted.
To the best of our knowledge, the TDH monomer has not previously been employed in the synthesis of polyurea.
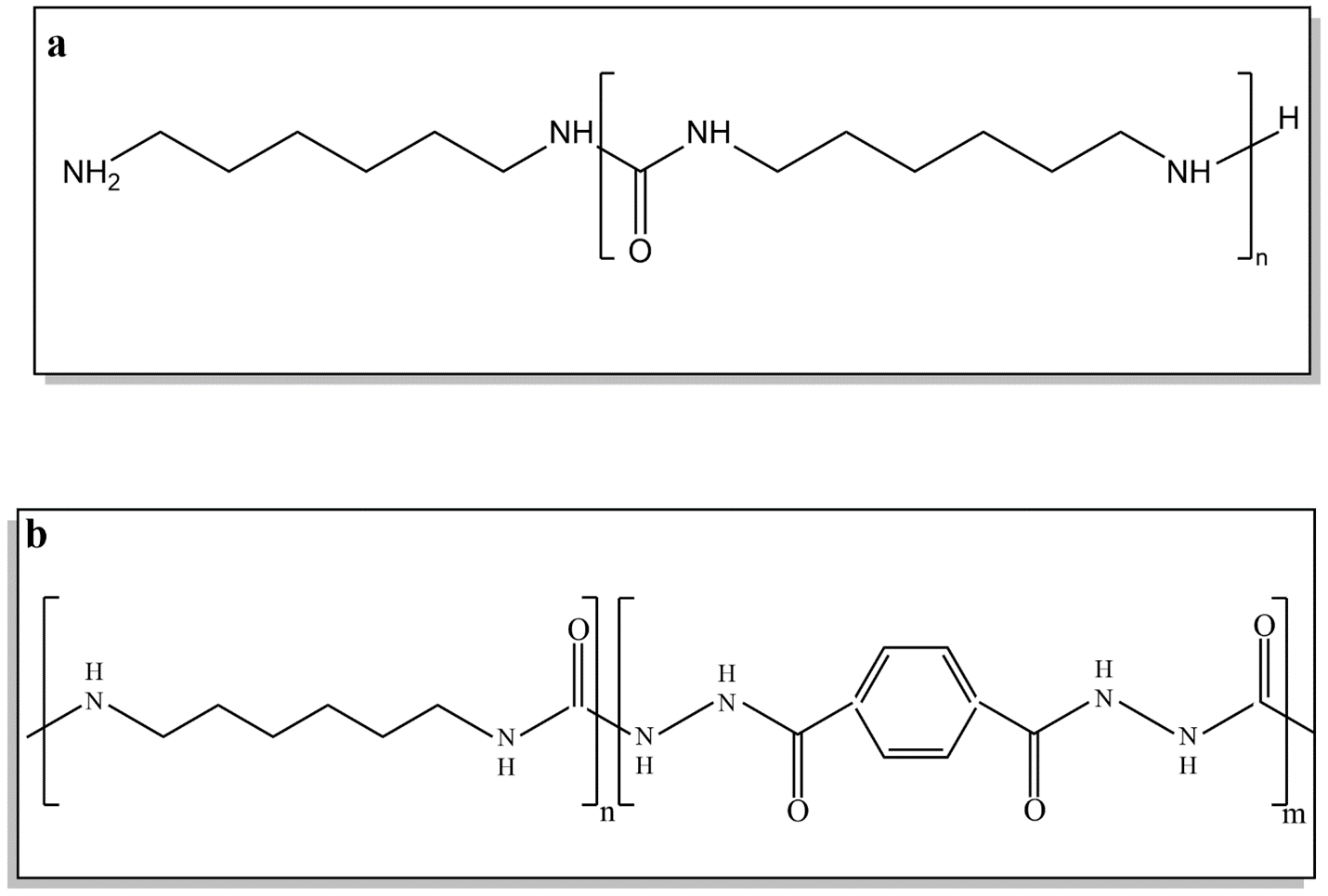
In this research, we synthesized both copolyurea and homopolyurea(scheme 3). The data and results from the homopolymer were used as a basis for developing copolyureas that incorporate TDH.For this purpose, HDA100 samples were prepared within a pressure range of 9 to 12 MPa, a temperature range of 160 to 190 degrees Celsius, and a reaction time of 6 to 12 h. Considering the influence of the three parameters—temperature, pressure, and time—on reaction efficiency, and the need to achieve optimal conditions, each series of samples was tested by keeping two of the parameters constant while varying the third. The optimal value for each parameter was thus determined. To estimate the reaction yield, the weight ratio of the product (Wp) to the initial feed (Wi) was calculated for various HDA100 samples.
According to the results of the reaction yield in the first stage of the synthesis, the optimum temperature, time, and pressure were determined to be 180°C, 8 h, and 11 MPa, respectively. These optimal conditions for the HDA100 have been used as the basis for the synthesis of copolymers. For the first stage of copolymer synthesis, the optimal conditions were 210°C, 10 h, and 11.5 MPa, respectively. Based on our initial observations during the synthesis of TDH-containing copolymers, samples with high percentages of the hard segment (TDH), specifically 60%, 70%, 80%, 90%, and 100%, were found to be unprocessable. Consequently, we opted against prototyping or synthesizing compounds exceeding a 50% hard segment composition. This decision resulted in setting the maximum allowable TDH content in the copolymers at 50%.
After determining the optimal values for the key factors influencing the reaction during the oligomerization stage, the second synthesis stage was initiated. In this phase, three main parameters must be assessed to establish their optimal values. Additionally, considering the synthesis reaction of the second stage in a three-neck glass balloon and the limitation on applied pressure, all polyurea samples at this stage were synthesized under an applied pressure of 0.1 MPa, as suggested by existing literature and reports. Observations also indicated that a total duration of 2 h for the post-polymerization reaction (t2nd and t3rd) was the optimal time for conducting the reaction. In light of the cases mentioned and considering the composition of the desired polymer for synthesis, the focus was solely on the temperature parameter (T2nd and T3rd). Generally, during sample preparation, the molar percentage of the hard segment in the formulation was incrementally raised, and based on these adjustments, the suitable temperatures were assessed. Table 4 displays the optimal values for the polyurea synthesis process.
Table 4
the optimal values for the synthesis process of both homopolymer and copolymer polyureas
| Entry | Sample | P1st (MPa) | T1st (°C) | t1st (h) | P2nd (MPa) | T2nd (°C) | t1st (h) | T2nd (°C) | t1st (h) |
| 1 | HDA100 | 11 | 180 | 8 | 0.1 | 200 | 1 | 250 | 1 |
| 2 | HDA90TDH10 | 11.5 | 210 | 10 | 0.1 | 200 | 1 | 250 | 1 |
| 3 | HDA80TDH20 | 11.5 | 210 | 10 | 0.1 | 210 | 1 | 260 | 1 |
| 4 | HDA70TDH30 | 11.5 | 210 | 10 | 0.1 | 220 | 1 | 270 | 1 |
| 5 | HDA60TDH40 | 11.5 | 210 | 10 | 0.1 | 230 | 1 | 270 | 1 |
| 6 | HDA50TDH50 | 11.5 | 210 | 10 | 0.1 | 230 | 1 | 270 | 1 |
The structure of the synthesized polyureas was analyzed using FTIR spectroscopy. The results are shown in Fig. 7 for the polyurea oligomer and copolymer samples. The FTIR spectrum graphs of these samples reveal five distinct peaks within the tested wavelength range. Notably, the peak at 3334 cm–1 is attributed to the stretching vibration of the N–H group in the oligomer.[9, 11, 12] This peak shows a redshift of about 66 cm–1 from the free N–H group peak at 3450 cm–1, indicating the presence of intermolecular hydrogen bonds in the synthesized polyurea. Research by Jiang and colleagues on polyurea synthesis via direct polymerization of 1,6 hexane diamine and carbon dioxide also reported a similar N–H stretching vibration peak at 3329 cm–1.[12] Moreover, in studies aiming to synthesize thermoplastic polyureas using 1,12-aminododecane (DAD) and/or 4,7,10-trioxa-1-13-tridecane diamine (TTD) with carbon dioxide, they observed this N–H stretch at 3338 cm–1.[9]
In separate research led by Ruhui Shi and his team, the stretching vibration of the N–H group was detected at a wave number of 3429 cm–1.[11]
The FTIR spectra also highlight a peak at 1619 cm–1, associated with the stretching vibration of the –C(= O)– group in the synthesized samples. This peak exhibits a shift of approximately 70 cm–1 to a lower wavelength compared to its free state at 1690 cm–1, which suggests the formation of hydrogen bonds. According to literature in the field, the –C(= O)– peak for oligomer samples typically ranges from 1618 to 1614 cm–1, and for polyurea from 1630 to 1622 cm–1.[8]
Additionally, the absorption peaks at 2930 cm–1 and 2857 cm–1 are identified as related to the stretching vibration of the C–H group along the main aliphatic chain. Jiang and colleagues reported similar absorption peaks at 2863 cm–1 and 2932 cm–1, while another study reported them at 2850 cm–1 and 2922 cm–1. Some studies also mention a bending vibration of the N–H group in the urea –CO–N–H– bonding group at 1573 cm–1. In this research, this peak appears independently in some homopolyurea and copolyurea samples, while in others, it overlaps with the C = O stretching vibration peak.
Based on the FTIR spectra of the synthesized samples and a comparison with existing literature, the formation of the urea functional group in the synthesized product can be confirmed.
Additionally, the chemical compositions of the synthesized polyurea were analyzed using 1HNMR, as shown in Fig. 8. 1H NMR results (Fig. 8) show that the actual molar ratio of HDA to TDH in the sample HDA80TDH20 is 4, corresponding to the initial molar ratio used in the feed.
This consistency is also seen in the HDA100 and HDA90TDH10 samples, based on their 1HNMR results. However, for samples with a hard segment content of 30–50%, such as HDA70TDH30, the peak distinction is compromised due to poor solubility in DMSO solvent (Fig. 9), resulting in fewer observable peaks. Further, the molecular weight and ratios of monomers in the final product can be assessed by comparing the integrated areas of characteristic peaks. The discrepancies between the initial feed ratios and the 1HNMR results, particularly in samples with a higher hard segment content, are attributed to the incomplete dissolution in DMSO and some TDH monomers not reacting under the experimental conditions. According to the NMR data, the number-average molecular weight (Mn) for the HDA80TDH20 sample is approximately 5600 g/mol, while the Mn for the HDA70TDH30 oligomer is around 3600 g/mol.
The thermal properties of the prepared polyureas, HDAxTDH100–x, were analyzed using DSC and TGA. The DSC curves from the second heating run can be found in Fig. 6a. The data reveal that the melting temperatures (Tm) for HDA100, HDA90TDH10, and HDA80TDH20 are 269°C, 244°C, and 238°C, respectively. Additionally, the melting temperature of the homopolymer HDA100 aligns with the findings from previous research conducted by other scholars.[12]
The decrease in melting temperature observed in this series of polyurea samples indicates that as the HDA content decreases, or conversely, as the TDH content increases, the melting temperature drops by approximately 30°C.
However, according to Fig. 10a for the other samples HDA70TDH30, HDA60TDH40, and HDA50TDH50, the melting peak disappears as the TDH content increases in the copolymer structure. It can be concluded that the increase in TDH reduces crystalline structure, thereby promoting the formation of an amorphous structure. Figure 10b illustrates the thermal behavior during the DSC cooling process, showing that the crystallization temperature (Tc) undergoes a similar alteration with the addition of TDH to HDA.
Figure 10b demonstrates a trend in the crystallization temperatures of the samples similar to that observed in their melting temperatures. Specifically, the crystallization temperatures for HDA100, HDA90TDH10, and HDA80TDH20 are 200°C, 189°C, and 181°C, respectively. It is inferred that an increase in the TDH fraction within the copolyureas leads to a decrease in crystallinity.
Table 5 provides the thermal characteristics of synthesized polyurea samples, including melting enthalpy (ΔHm), crystallization enthalpy (ΔHc), melting temperature (Tm), crystallization temperature (Tc), and the initial decomposition temperature at 5% weight loss (Td, 5%).
Given the influence of molecular weight, degree of polymerization, and molecular structure on polymers’ thermal properties and stability, a TGA (Thermogravimetric Analysis) test was employed to study the structure of homopolyurea copolyureas.
Figure 11 presents the spectra from the thermal gravimetric tests, which compare the thermal decomposition processes of the synthesized polyurea samples.
Figure 11 shows that the homopolymer sample's initial decomposition temperature (Td, 5%) is 217°C. Increasing the content of the TDH monomer in the copolymer structure, which includes an aromatic ring as a hard segment, results in higher thermal stability in the copolymer samples compared to the homopolymer. According to the TGA curve in Fig. 11, the initial decomposition temperatures of the copolyurea samples for HDA60TDH40 and HDA50TDH50 are approximately 336°C and 366°C, respectively. In samples with a lower TDH or higher HDA content, the increase in the initial decomposition temperature is less pronounced compared to the homopolymer. For example, the initial decomposition temperatures for HDA90TDH10, HDA80TDH20, and HDA70TDH30 are 220°C, 235°C, and 285°C, respectively.
Table 5
Thermal properties of polyureas
| | | DSC | TGA |
| Entry | Sample | Tm (°C) | Tc (°C) | ΔHm (J/g) | ΔHc (J/g) | Td,5% (°C) |
| 1 | HDA100 | 269 | 200 | 90 | 50 | 217 |
| 2 | HDA90TDH10 | 244 | 189 | 57 | 48 | 220 |
| 3 | HDA80TDH20 | 238 | 181 | 53 | 45 | 235 |
| 4 | HDA70TDH30 | - | - | - | - | 285 |
| 5 | HDA60TDH40 | - | - | - | - | 336 |
| 6 | HDA50TDH50 | - | - | - | - | 366 |
{kind=link}
{kind=link}
{kind=link}