Materials: DMEM/F12 (HyClone, USA), Penicillin and streptomycin (Gibco, Australia), Fetal bovine serum (FBS, HyClone, USA), trypsin containing 0.02% EDTA (Sigma–Aldrich, St. Louis, USA), Cell Counting Kit-8 (CCK-8, BBI life Science, Sangon Biotech, China), PBS-EDTA (P885743, Macklin, China), RADA16 peptide (RADARADARADARADA),TR peptide (HSNGLPL-GSG-RADARADAR
ADARADA), RM peptide (RADARADARADARADA-GSG-SKPPGTSS), TRM peptide (HSNGLPL-GSG-RADARADARADARADA-GSG-SKPPGTSS) was synthesized by China Peptides Co, Ltd. (Shanghai, China), active TGF-β1 (ab50038, Abcam, Cambridge, UK), TGF-β1 ELISA Kit (YX-200709, Sinobestbio, China), Crystal Violet Staining Solution (C0121, Beyotime Biotechnology, China), Actin-Tracker Red-Rhodamine (C2207S, Beyotime Biotechnology, China), 4′,6-diamidino-2-phenylindole (DAPI, C1006, Beyotime Biotechnology, China), Immunol Staining Blocking Buffer (P0102, Beyotime Biotechnology, China), Tissues Embedding Medium-optimal cutting temperature (OCT) compound (4583, Sakura Finetechnical Co Ltd., Tokyo, Japan), antibody anti-SOX9 (ab185230, Abcam, 1:200, Cambridge, UK), antibody anti-TGF-β1 (ab215715, Abcam, Cambridge, UK), antibody anti-CD105 (ab156756, Abcam, Cambridge, UK), antibody anti-type II collagen (ab188570, Abcam, Cambridge, UK), Alexa Fluor 488-conjugated secondary antibody (ab150077, Abcam, Cambridge, UK), Alexa Fluor 647-conjugated secondary antibody (ab150115, Abcam, Cambridge, UK). RNAiso Plus (15596026, Invitrogen, California, USA), iTaq Universal SYBR Green Supermix (1725121, Bio-Rad, Hercules, CA, USA), RevertAidTM First Strand cDNA Synthesis Kit (K16225, Thermo Fisher, MA, USA).
Preparation of Exsomes
In 10 cm cell culture dishes, 6× 106 BMSCs cells were planted. Next, the medium was collected, and cell debris were removed by 1000 g for 10 min and 14,000 g for 2 min. exsomes were gained from the supernatant via 100,000 g for further 60 min at 4°C and washed with sterile 1×PBS for 2 times. At last, the exsomes were resuspended with 1×PBS for subsequent experiments.
Quantifcation of Exsomes
The quantifcation of exsomes were determined by their protein concentrations. Radioimmunoprecipitation assay bufer were applied to lyse exsomes at 4°C for half an hour. Then, the lysis was centrifuged at 12,000 g for 30 min at 4°C and the supernatant was transferred into a new centrifuge tube for protein concentration measurement by BCA Protein Assay Kit (Termo Fisher Scientifc).
Characterization of Exsome Size and Transmission electron microscopy (TEM)
One milliliter of 30 ng mL− 1 exsomes were taken for the measurement of the particle size and polydispersity index by Malvern laser particle size analyzer (Zetasizer Nano ZSP). For further identifcation of the sizes and morphology of exsomes were washed by ddH2O, deposited on copper mesh and then observed by TEM (HT7700-SS/FEI Tecnai G20 TWIN).
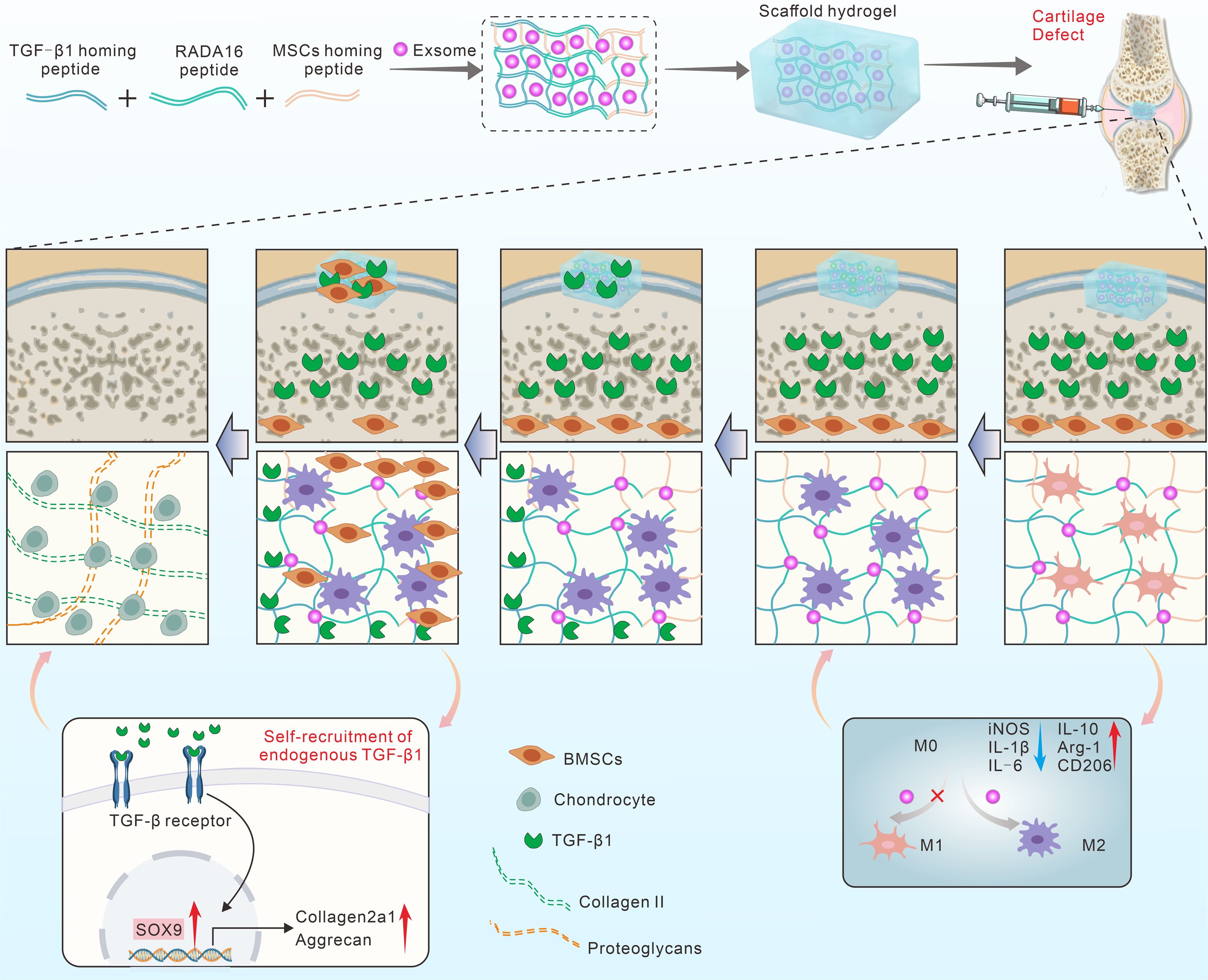
Hydrogel Preparation: To synthesis of indicated peptide hydrogel, 10 mg of RADA16 peptide or equimolar concentrations of other fusion peptides and exosomes (1mg total protein) was dissolved in an aqueous solution containing 0.9% sodium chloride and incubated overnight at 4°C to obtain the aqueous gel system for the experiment.
Surface morphology: The surface morphologies of the hydrogels were determined using scanning electron microscopy (SEM, SU-8010, Japan) equipped with an energy dispersive spectroscopy (EDS). Samples were freeze-dried, and the cross-sections of the samples were coated with a gold layer to enhance the conductivity for SEM.
Release of Exosome and peptide from hydrogels: 200 µL hydrogel was immersed in 2 mL of phosphatebuffered saline (PBS) containing 5 unit/mL proteinase K and incubated at 37℃ to investigate the release kinetics of peptide and exosomes from the indicated hydrogel. Supernatant (200 µL) was collected from the tube at specified time intervals, and an equal volume of fresh PBS was replenished. The concentration of exosomes and peptide in the supernatant was determined by measuring the OD at 265 nm using a UV spectrophotometer (MIULAB, China) and employing a bicinchoninic acid (BCA) protein assay kit (Beyotime, China) to quantify the protein content. Finally, the cumulative release of peptide and exosomes from the indicated hydrogel was calculated.
Uptake of Exos: The phagocytosis of exosomes encapsulated in the hydrogel or alone by BMSCs was investigated as follows. We first performed exosomes isolation, identification and staining. Exosomes were initially labeled with PKH 26 and subsequently co-incubated with BMSCs for 24 h. The cells were then fixed at room temperature and stained with TRITC-phalloidin (Invitrogen, USA) and 4-6-diamidino-2-phenylindole (DAPI, Invitrogen) to identify the cytoskeleton and nucleus, respectively. Finally, labeled cells were observed using an LSM710 laser scanning confocal microscope.
In vitro biocompatibility assessment: The cytocompatibility of the hydrogel or exosomes were evaluated using a live/ dead staining kit and CCK-8 assay kit. For live/dead staining, BMSCs were cultured with the hydrogel or exosomes for 1, 3, and 7 days. Staining was conducted per manufacturer’s instructions, and the cells were observed using an LSM710 laser scanning confocal microscope. For the cyto-toxicity assay, BMSCs were co-cultured with the hydrogel or exosomes for 1, 3, and 7 days. Subsequently, 100 µL of CCK-8 solution (100 µL/mL) was added to each well, and incubation continued for 2 h. A microplate reader (Tecan Spark, Switzerland) was used to measure the absorbance of cells in each well at 450 nm.
Flow cytometry analysis of macrophage polarization: BMDM cells were co-cultured with the exosomes or indicated hydrogel in well plates and stimulated with either LPS (100 ng/mL) or IL-4 (20 ng/mL). Afterwards, the cells were washed with pre-cooled PBS to prepare single-cell suspensions. This was followed by a series of steps, including fixation, membrane-breaking, and incubation with antibodies targeting M1 marker CD86 and M2 marker CD206 (Biolegend). Subsequently, the cells were analyzed using a flow cytometer and analyzed using FlowJo.
Gene expression: Total cellular RNA was determined using an RNA extraction kit (Omega, USA). cDNA synthesis was performed using a PrimeScript RT reagent kit (Takara, Japan), and qPCR amplification was conducted using the qPCR SYBR green master mix (Yeasen, China). The GAPDH gene was used as an internal control. Relative gene expression levels were measured using the 2−ΔΔCt method. The expressions of inflammation-related genes (IL-1β, IL-6, IL-10, Arg-1), were evaluated. The experiments of real-time quantitative PCR (qPCR) were repeated three times independently. Primers for this study were purchased from Tsingke Biotech (China), and the sequences are listed as follows:
Table 1
Primer sequences of each gene
| Target | Forward | Reverse |
| IL-1β | CAGCACATCAACAAGAGCTTCAG | GAGGATGGGCTCTTCTTCAAAGA |
| IL-6 | GAGTCCTTCAGAGAGATACAG | CTGTGACTCCAGCTTATCTG |
| Arg-1 | TCTGCCAAAGACATCGTGTACAT | CGACATCAAAGCTCAGGTGAATC |
| IL-10 | CTGCTCTTACTGGCTGGAGTGAAG | TGGGTCTGGCTGACTGGGAAG |
| GAPDH (Rat) | CAGTGGCAAAGTGGAGATTGTTG | TCGCTCCTGGAAGATGGTGAT |
Immunofluorescence: After the indicated treatment, the cartilage sites of knee joints were obtained from rat, and fixed in 4% paraformaldehyde for 24 h, and then dehydrated in 30% (m/V) sucrose solution for 48 h. Cryosections (10 µm) were cut using a cryostat (Leica, CM1950). The slices were blocked with blocking buffer (3% (m/V) BSAand 0.2% (m/V) Triton X-100 in PBS) for 1 h Samples were incubated with primary antibodies overnight at 4 ℃. Subsequently, secondary antibodies were applied, and nuclei were stained with DAPI (BD Biosciences, cat: 564907) at room temperature for 5 min. After washing in PBS, the slices were mounted and imaged using spinning-disk confocal microscopy (PerkinElmer Instrument Co., Ltd., Germany).
Western blotting: The cells were collected and added to RIPA lysis buffer (Beyotime)
containing protease and phosphatase inhibitors (Sigma-Aldrich). Then, the solution was centrifuged at 12000 rpm for 30 min at 4℃ to isolate the supernatant. SDS-PAGE was used to separate the proteins, which were transferred onto 0.45 µm PVDF membrane (Millipore, USA), blocked with QuickBlockTM blocking buffer (Beyotime), and then incubated with primary antibodies (listed in Table S2) at 4℃ overnight. Finally, the proteins were incubated with secondary antibodies (Proteintech, China) at room temperature for 2 h. An enhanced chemiluminescence kit (Millipore) was applied to detect the proteins.
Rat cartilage defect model: All the animals were treated according to the standard guidelines approved from the Animal Ethics Committee of Shandong Provincial Hospital Affiliated to Shandong First Medical University (No. 2023 − 178). Adult male SD rats (10 weeks old) were used in the in vivo study. After anesthesia, a cylindrical osteochondral defect of 2 mm in diameter and 2 mm in depth was created on the patellar groove. Rats were randomized into 4 groups: Sham (n = 10), Defect (n = 10 joints), TRM peptide hydrogel (n = 10), Exosomes (n = 10) and Exos@TRM (n = 10). In the defect group, the defects were untreated. The other treatment group were injected into the defects, respectively. After the operation, the rats were fed with standard food and water, and penicillin was injected intramuscularly to prevent infection.
Endogenous TGF-β1 Recruitment and BMSCs Infiltration In vivo: At day 14 post-surgery, 3 knees of each group were collected and fixed with 4% paraformaldehyde for 24 h and decalcified with 10% EDTA for one month. The samples were embedded in an OCT compound and sectioned into 15 µm through the center of the graft. In the detection of endogenous TGF-β1 recruitment, the samples were incubated with primary antibodies, including anti-TGF-β1 (1:200) and counterstained with DAPI. In the detection of endogenous BMSCs infiltration, the samples were incubated with primary antibodies, including anti-CD105 (1:200) and counterstained with DAPI.
Histological Analysis
The knee joints were fixed with 4% paraformaldehyde for 24 h and decalcified with 10% EDTA for one month. The samples were embedded in paraffin and sectioned into 15 µm through the center of the graft. Safranin O/fast green staining was used to evaluate the osteo-chondral repair, while immunofluorescence by type II collagen was used to analyze cartilage matrix deposition. All images were captured by an LSM710 laser scanning confocal microscope.
Statistical analysis
All data are expressed as the mean ± standard deviation (n ≥ 3). Variations between multiple groups were assessed using one-way analysis of variance (ANOVA). Results were analyzed and compared using GraphPad Prism software (LaJolla, CA). Statistical significance was set at ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
{kind=link}